

The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations
- PMID: 28481356
- PMCID: PMC5737966
- DOI: 10.1038/nsmb.3408
The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations
Abstract
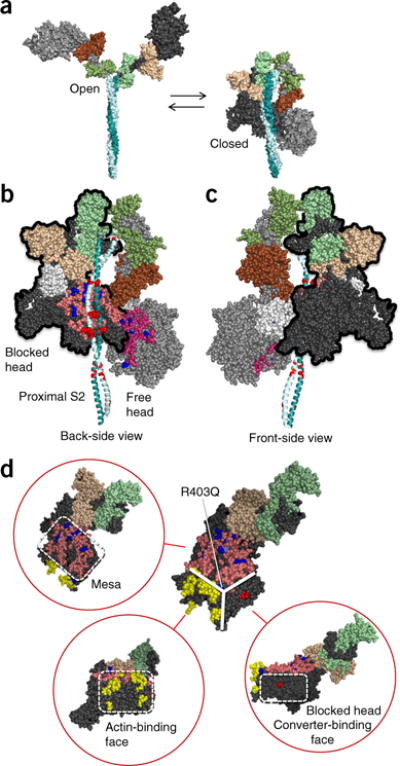
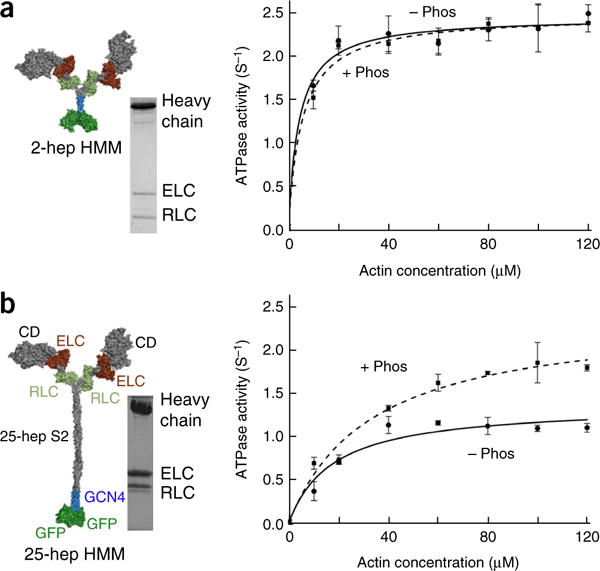
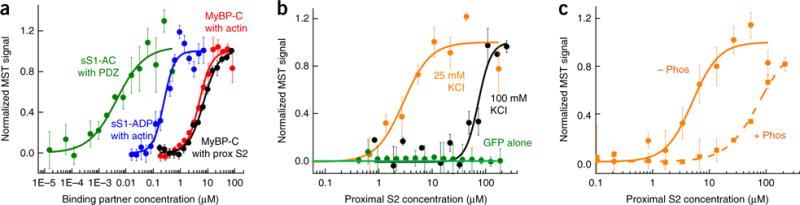
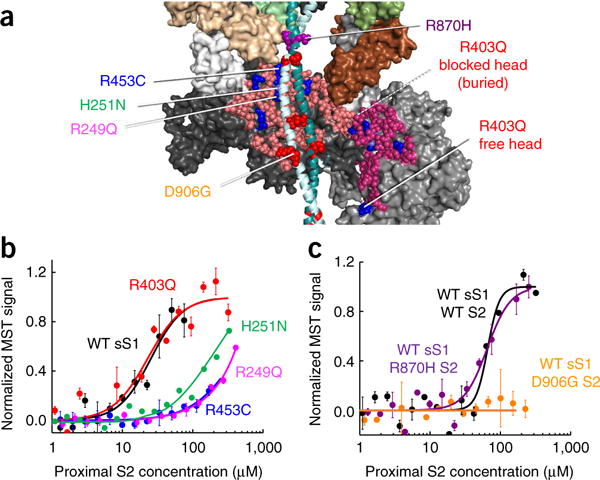
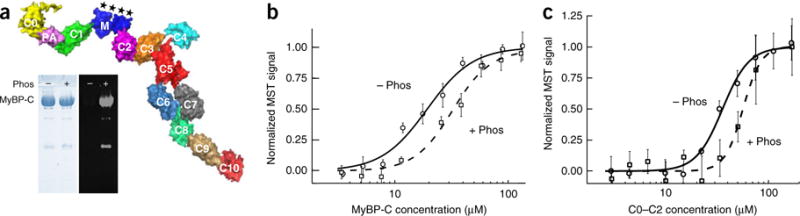
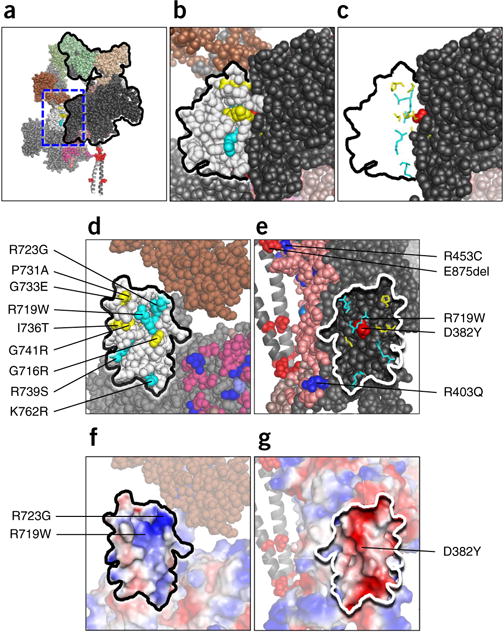
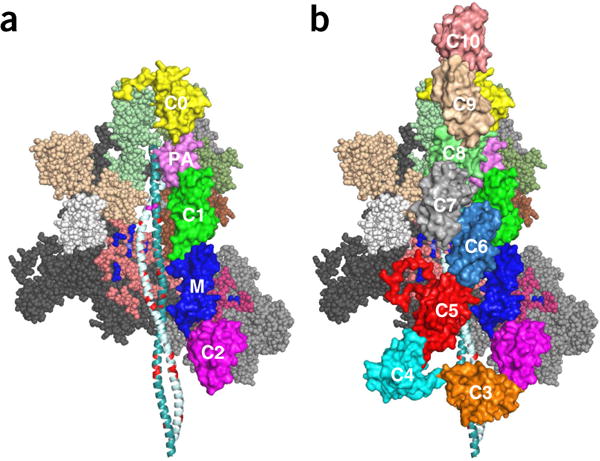
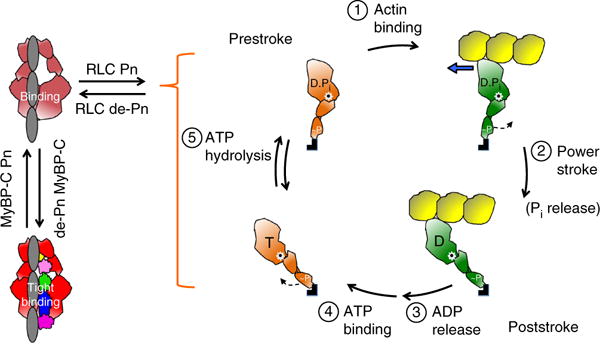
Hypertrophic cardiomyopathy (HCM) is primarily caused by mutations in β-cardiac myosin and myosin-binding protein-C (MyBP-C). Changes in the contractile parameters of myosin measured so far do not explain the clinical hypercontractility caused by such mutations. We propose that hypercontractility is due to an increase in the number of myosin heads (S1) that are accessible for force production. In support of this hypothesis, we demonstrate myosin tail (S2)-dependent functional regulation of actin-activated human β-cardiac myosin ATPase. In addition, we show that both S2 and MyBP-C bind to S1 and that phosphorylation of either S1 or MyBP-C weakens these interactions. Importantly, the S1-S2 interaction is also weakened by four myosin HCM-causing mutations but not by two other mutations. To explain these experimental results, we propose a working structural model involving multiple interactions, including those with myosin's own S2 and MyBP-C, that hold myosin in a sequestered state.
Conflict of interest statement
The authors declare competing financial interests: details are available in the online version of the paper.
Figures
References
-
- Woodhead JL, et al. Atomic model of a myosin filament in the relaxed state. Nature. 2005;436:1195–1199. - PubMed
-
- Pinto A, Sánchez F, Alamo L, Padrón R. The myosin interacting-heads motif is present in the relaxed thick filament of the striated muscle of scorpion. J Struct Biol. 2012;180:469–478. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
