

Applications of NMR and computational methodologies to study protein dynamics
- PMID: 28483383
- PMCID: PMC5545153
- DOI: 10.1016/j.abb.2017.05.002
Applications of NMR and computational methodologies to study protein dynamics
Abstract
Overwhelming evidence now illustrates the defining role of atomic-scale protein flexibility in biological events such as allostery, cell signaling, and enzyme catalysis. Over the years, spin relaxation nuclear magnetic resonance (NMR) has provided significant insights on the structural motions occurring on multiple time frames over the course of a protein life span. The present review article aims to illustrate to the broader community how this technique continues to shape many areas of protein science and engineering, in addition to being an indispensable tool for studying atomic-scale motions and functional characterization. Continuing developments in underlying NMR technology alongside software and hardware developments for complementary computational approaches now enable methodologies to routinely provide spatial directionality and structural representations traditionally harder to achieve solely using NMR spectroscopy. In addition to its well-established role in structural elucidation, we present recent examples that illustrate the combined power of selective isotope labeling, relaxation dispersion experiments, chemical shift analyses, and computational approaches for the characterization of conformational sub-states in proteins and enzymes.
Keywords: Allostery; Chemical shift analysis; Conformational sub-states; Protein dynamics; Quasi anharmonic analysis; Relaxation dispersion.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
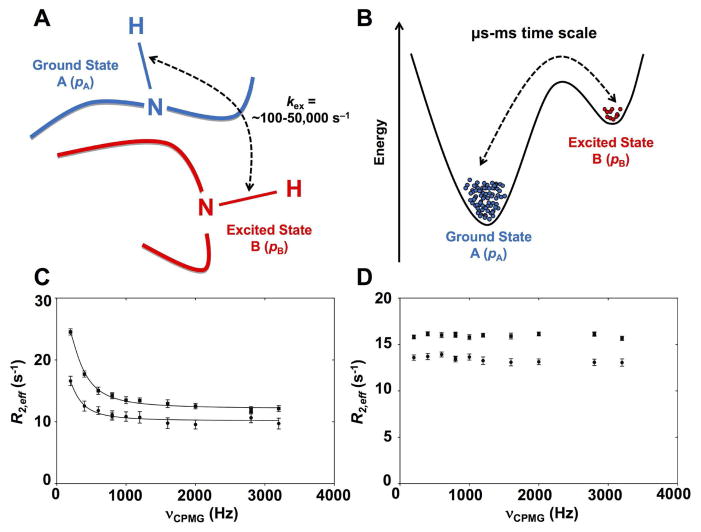
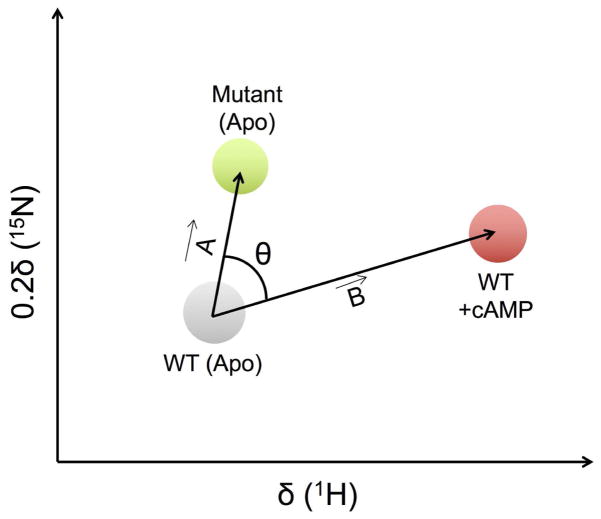
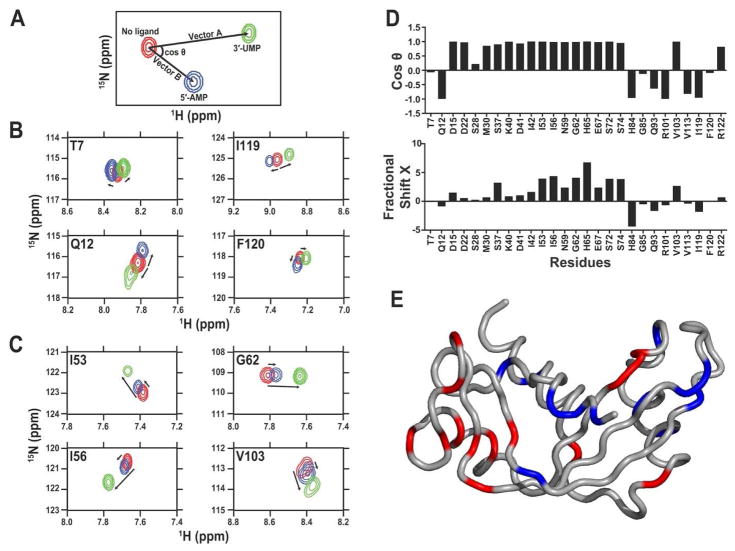
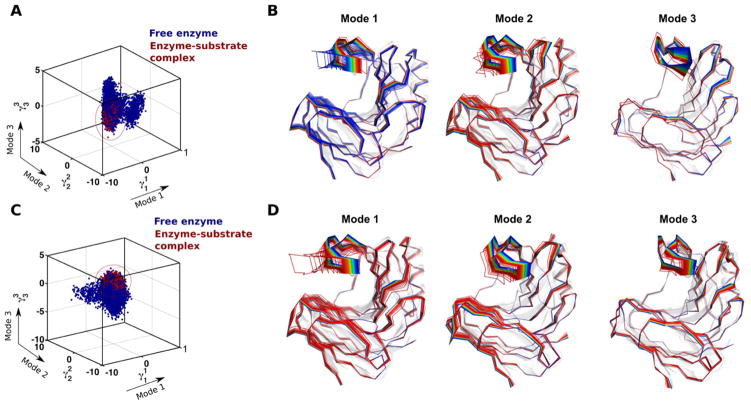
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
