

Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome
- PMID: 28483998
- PMCID: PMC5448167
- DOI: 10.1073/pnas.1621511114
Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome
Erratum in
-
Correction for Ferrari et al., Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome.Proc Natl Acad Sci U S A. 2017 Jun 13;114(24):E4894-E4896. doi: 10.1073/pnas.1708137114. Epub 2017 Jun 5. Proc Natl Acad Sci U S A. 2017. PMID: 28584118 Free PMC article. No abstract available.
-
Correction for Ferrari et al., Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome.Proc Natl Acad Sci U S A. 2018 Feb 6;115(6):E1330. doi: 10.1073/pnas.1800151115. Epub 2018 Jan 30. Proc Natl Acad Sci U S A. 2018. PMID: 29382743 Free PMC article. No abstract available.
Abstract
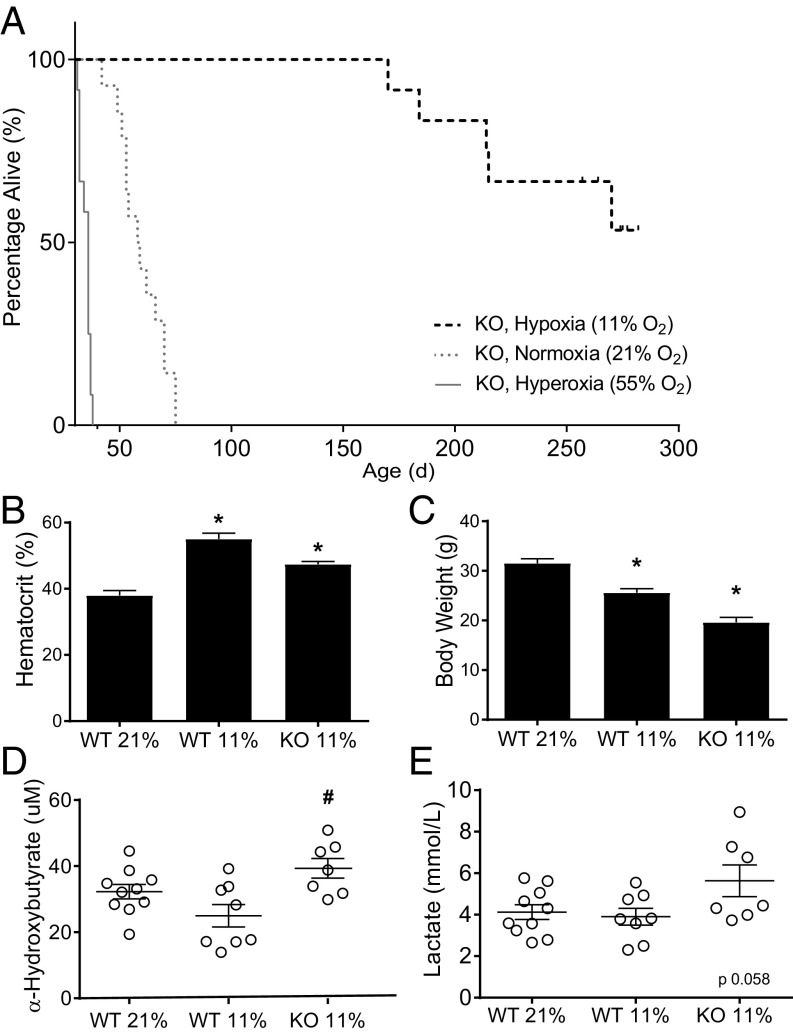
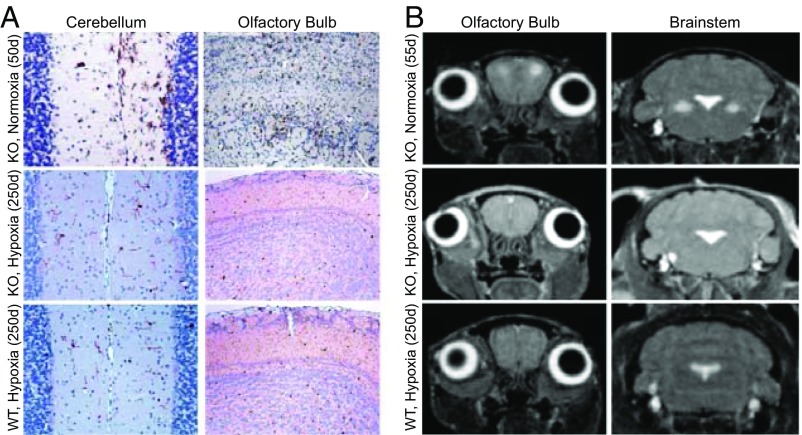
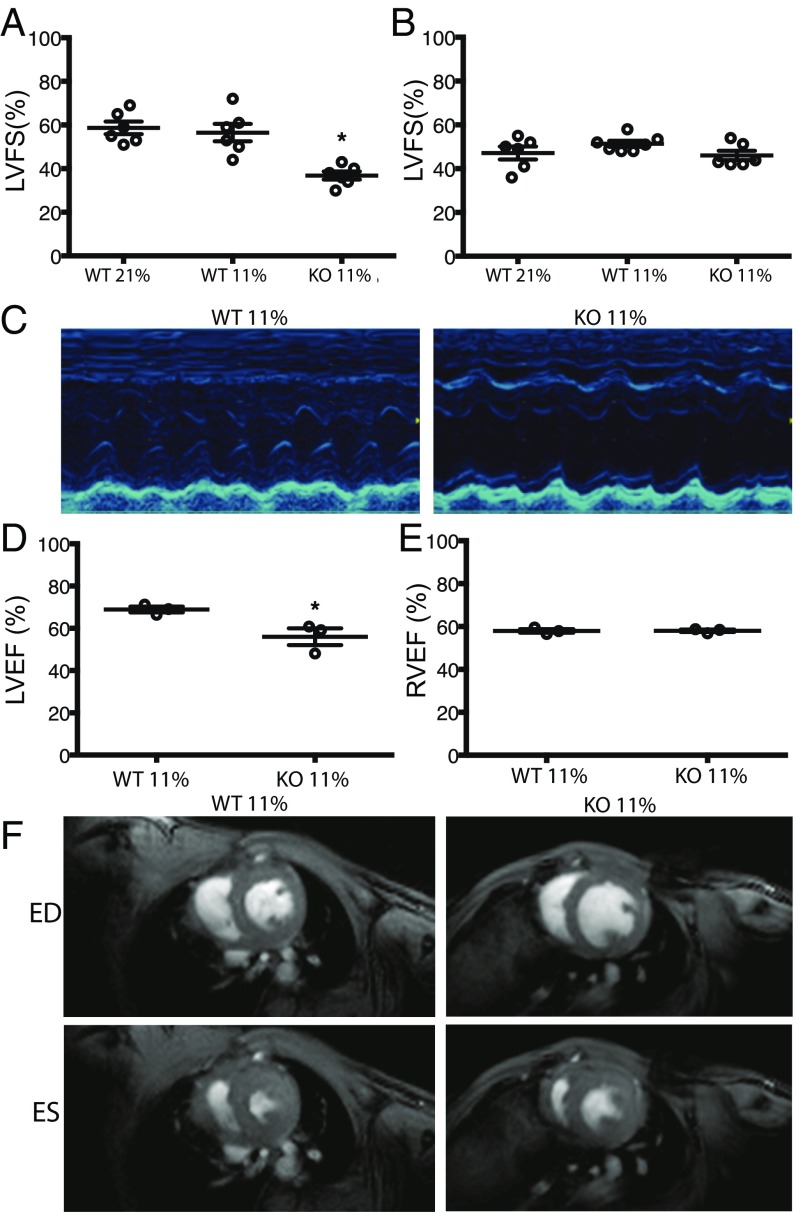
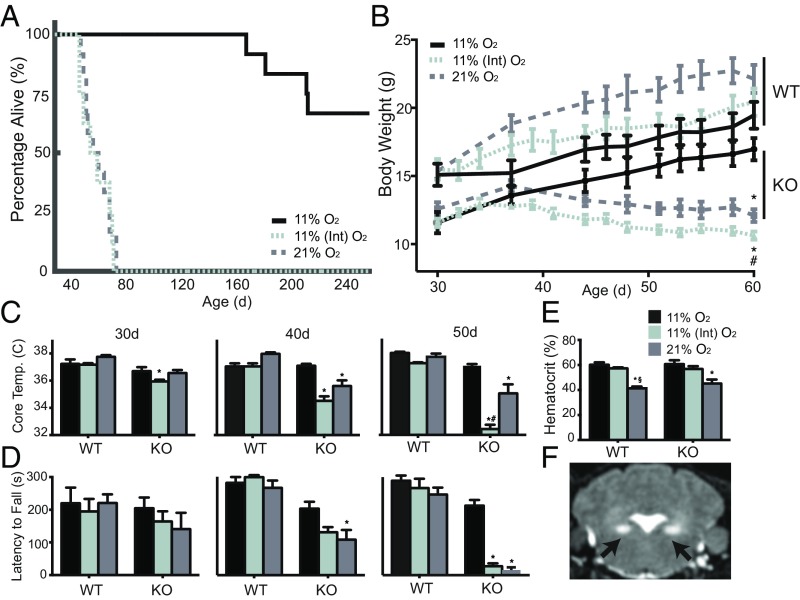
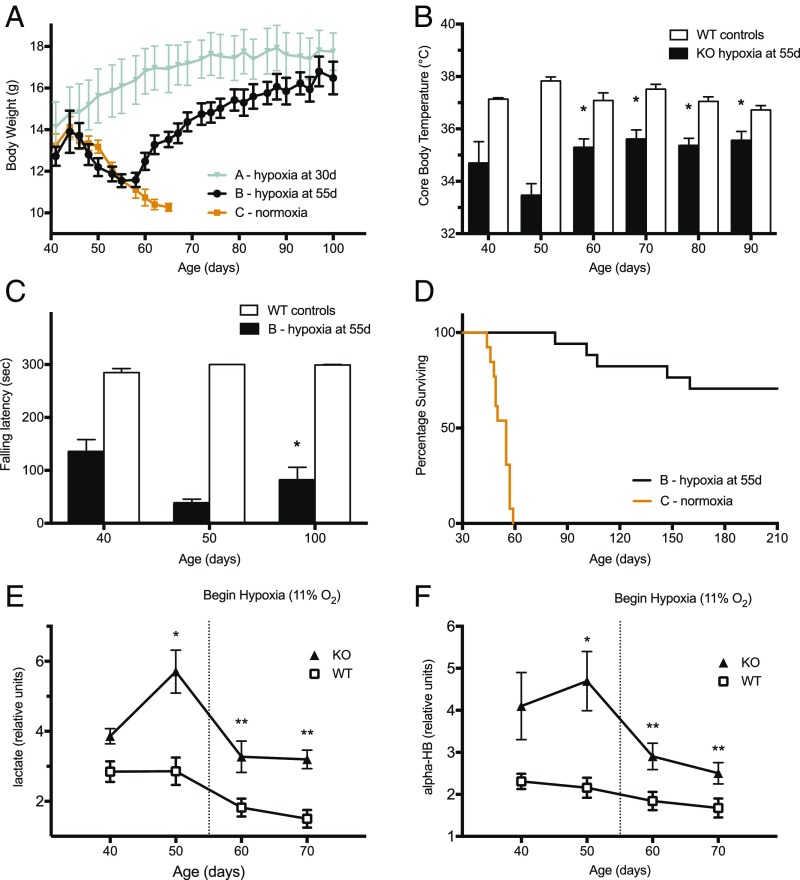
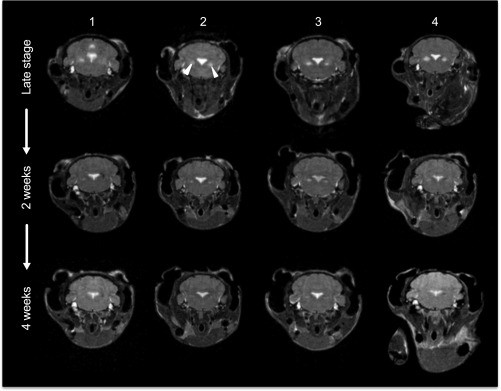
The most common pediatric mitochondrial disease is Leigh syndrome, an episodic, subacute neurodegeneration that can lead to death within the first few years of life, for which there are no proven general therapies. Mice lacking the complex I subunit, Ndufs4, develop a fatal progressive encephalopathy resembling Leigh syndrome and die at ≈60 d of age. We previously reported that continuously breathing normobaric 11% O2 from an early age prevents neurological disease and dramatically improves survival in these mice. Here, we report three advances. First, we report updated survival curves and organ pathology in Ndufs4 KO mice exposed to hypoxia or hyperoxia. Whereas normoxia-treated KO mice die from neurodegeneration at about 60 d, hypoxia-treated mice eventually die at about 270 d, likely from cardiac disease, and hyperoxia-treated mice die within days from acute pulmonary edema. Second, we report that more conservative hypoxia regimens, such as continuous normobaric 17% O2 or intermittent hypoxia, are ineffective in preventing neuropathology. Finally, we show that breathing normobaric 11% O2 in mice with late-stage encephalopathy reverses their established neurological disease, evidenced by improved behavior, circulating disease biomarkers, and survival rates. Importantly, the pathognomonic MRI brain lesions and neurohistopathologic findings are reversed after 4 wk of hypoxia. Upon return to normoxia, Ndufs4 KO mice die within days. Future work is required to determine if hypoxia can be used to prevent and reverse neurodegeneration in other animal models, and to determine if it can be provided in a safe and practical manner to allow in-hospital human therapeutic trials.
Keywords: Leigh syndrome; hypoxia; mitochondria; neurodegeneration; oxygen.
Conflict of interest statement
Conflict of interest statement: V.K.M., W.M.Z., and I.H.J. are listed as coinventors on a patent application submitted by Massachusetts General Hospital on the therapeutic uses of hypoxia for mitochondrial disease.
Figures



References
-
- Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383. - PubMed
-
- Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol. 2016;79:190–203. - PubMed
-
- Debs R, et al. Biotin-responsive basal ganglia disease in ethnic Europeans with novel SLC19A3 mutations. Arch Neurol. 2010;67:126–130. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
