

Genetic diagnosis as a tool for personalized treatment of Duchenne muscular dystrophy
- PMID: 28484312
- PMCID: PMC5416739
Genetic diagnosis as a tool for personalized treatment of Duchenne muscular dystrophy
Abstract
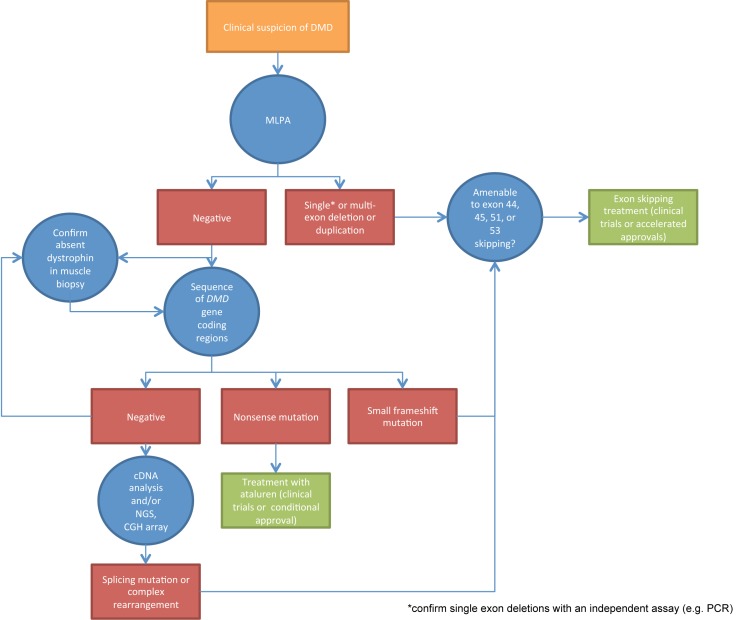
Accurate definition of genetic mutations causing Duchenne muscular dystrophy (DMD) has always been relevant in order to provide genetic counseling to patients and families, and helps to establish the prognosis in the case where the distinction between Duchenne, Becker, or intermediate muscular dystrophy is not obvious. As molecular treatments aimed at dystrophin restoration in DMD are increasingly available as commercialized drugs or within clinical trials, genetic diagnosis has become an indispensable tool in order to determine eligibility for these treatments. DMD patients in which multiplex ligation-dependent probe amplification (MLPA) or similar techniques show a deletion suitable to exon skipping of exons 44, 45, 51, or 53, may be currently treated with AONs targeting these exons, in the context of clinical trials, or, as is the case for exon 51 skipping in the United States, with the first commercialized drug (eteplirsen). Patients who test negative at MLPA, but in whom DMD gene sequencing shows a nonsense mutation, may be amenable for treatment with stop codon readthrough compounds such as ataluren. Novel molecular approaches such as CRISPR-Cas9 targeting of specific DMD mutations are still in the preclinical stages, but appear promising. In conclusion, an accurate genetic diagnosis represents the entrance into a new scenario of personalized medicine in DMD.
Keywords: CRISPR-Cas9; Duchenne muscular dystrophy; exon skipping; genetic diagnosis; stop codon readthrough.
Figures
References
-
- Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. - PubMed
-
- Hoffman EP, Fischbeck KH, Brown RH, et al. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne's or Becker's muscular dystrophy. N Engl J Med. 1988;318:1363–1368. - PubMed
-
- Ricotti V, Muntoni F, Voit T. Challenges of clinical trial design for DMD. Neuromuscul Disord. 2015;25:932–935. - PubMed
-
- Haas M, Vlcek V, Balabanov P, et al. European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul Disord NMD. 2015;25:5–13. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials