

Evolution of reduced co-activator dependence led to target expansion of a starvation response pathway
- PMID: 28485712
- PMCID: PMC5446240
- DOI: 10.7554/eLife.25157
Evolution of reduced co-activator dependence led to target expansion of a starvation response pathway
Erratum in
-
Correction: Evolution of reduced co-activator dependence led to target expansion of a starvation response pathway.Elife. 2022 Oct 20;11:e84187. doi: 10.7554/eLife.84187. Elife. 2022. PMID: 36264273 Free PMC article.
Abstract
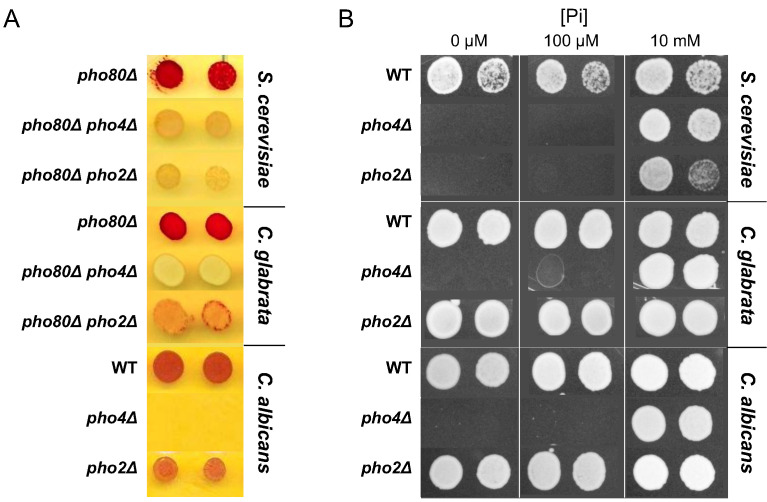
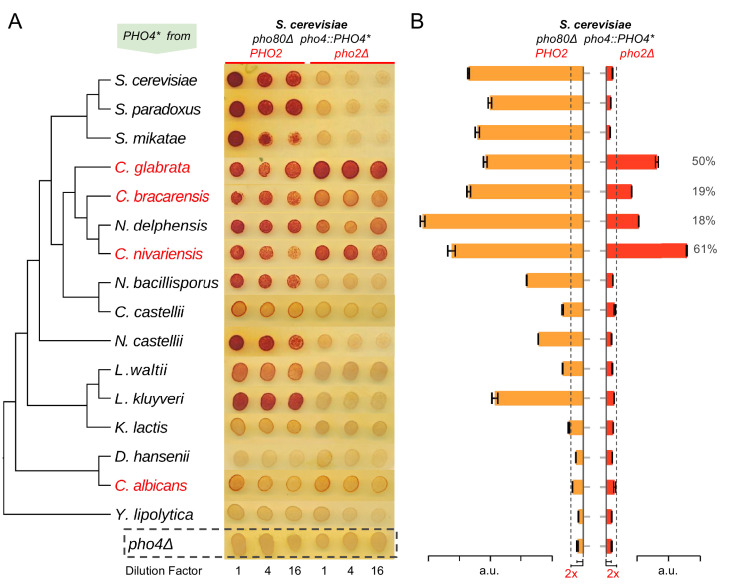
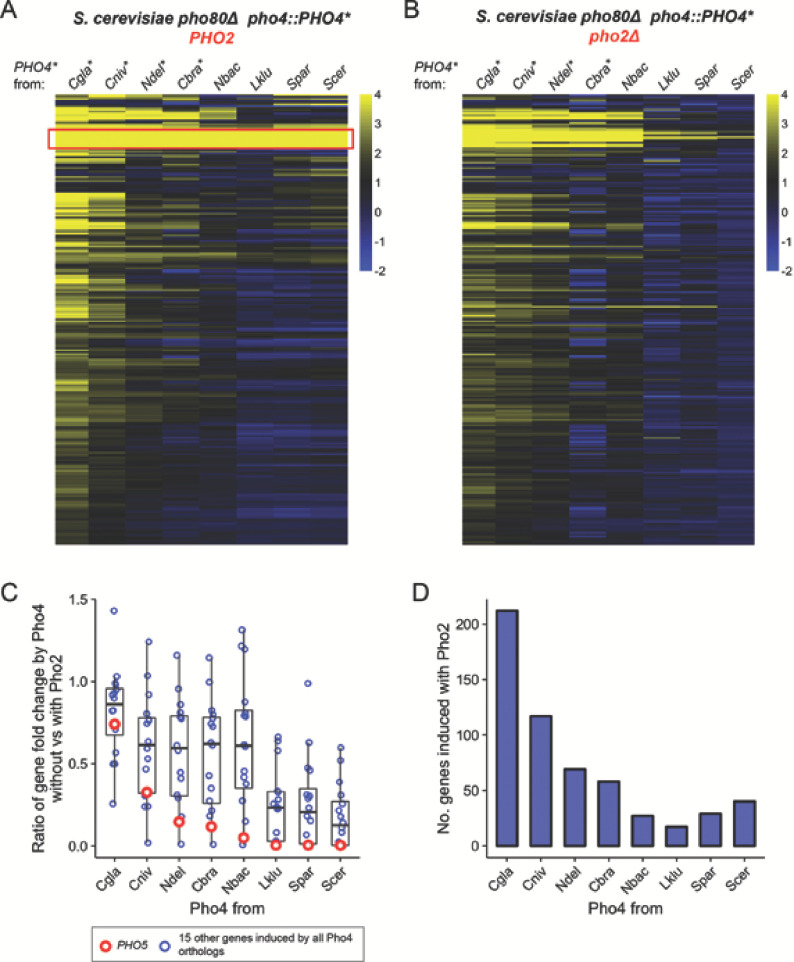
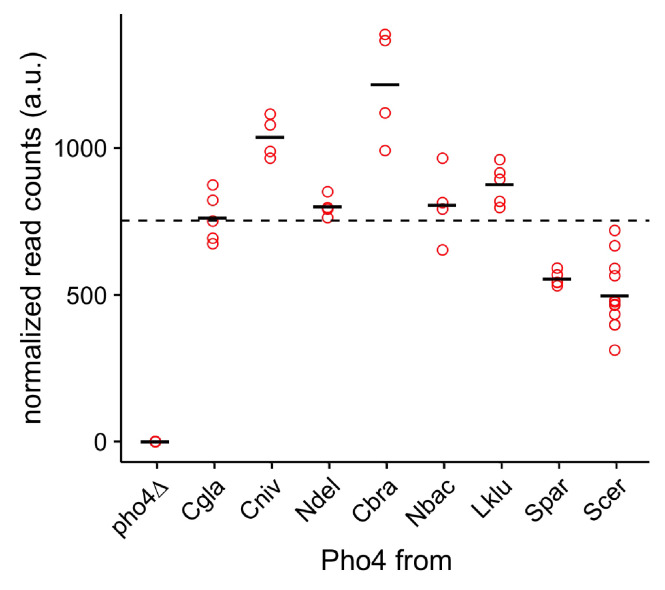
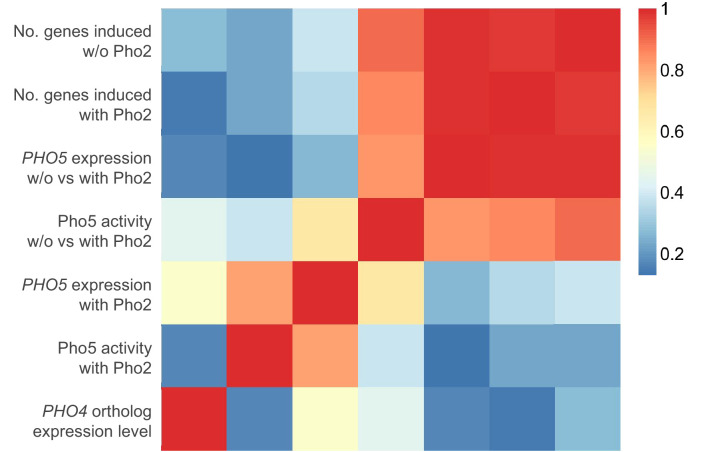
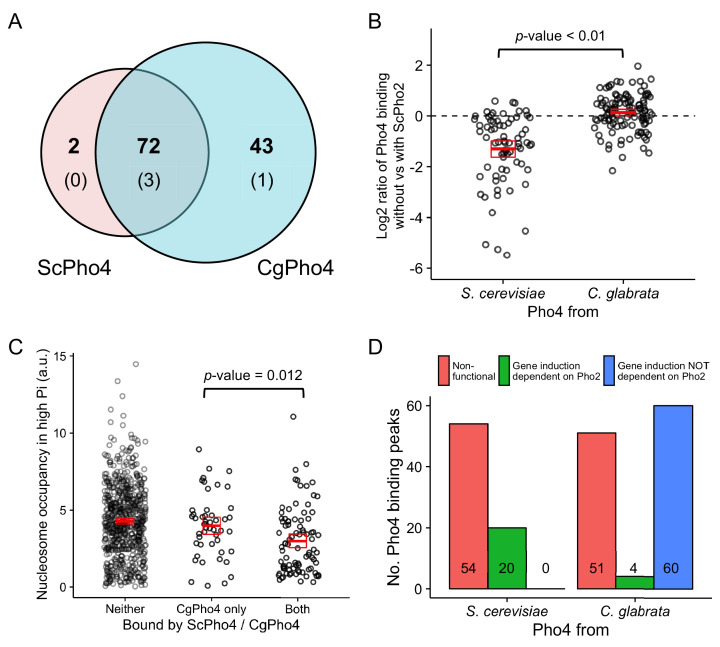
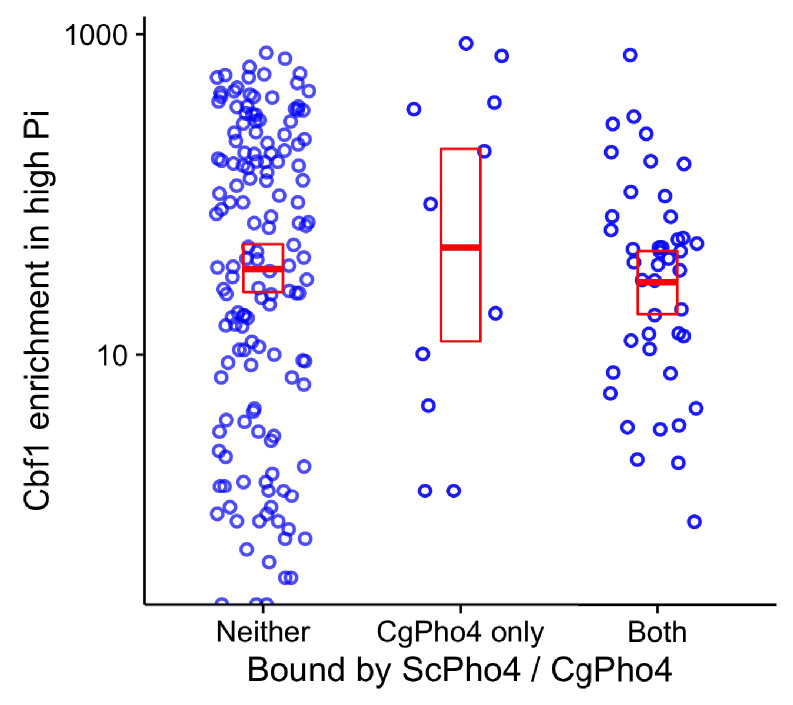
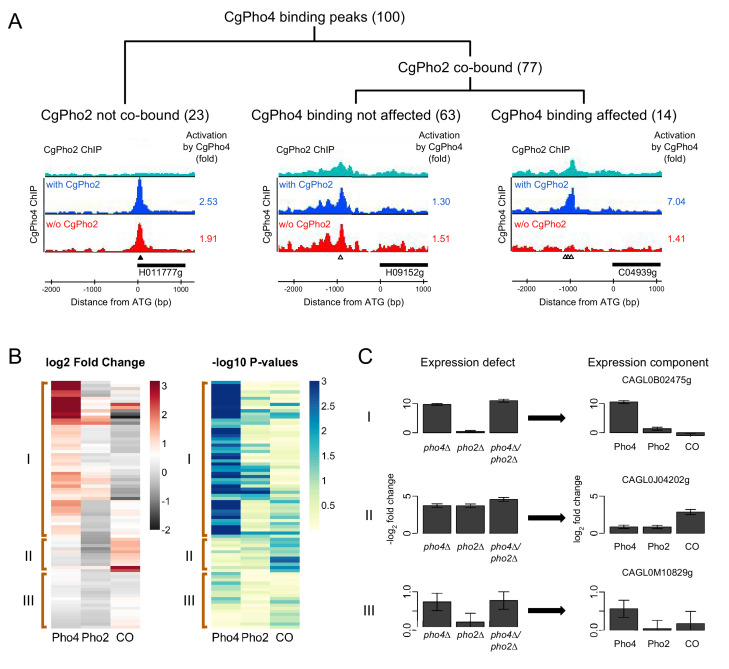
Although combinatorial regulation is a common feature in gene regulatory networks, how it evolves and affects network structure and function is not well understood. In S. cerevisiae, the phosphate starvation (PHO) responsive transcription factors Pho4 and Pho2 are required for gene induction and survival during phosphate starvation. In the related human commensal C. glabrata, Pho4 is required but Pho2 is dispensable for survival in phosphate starvation and is only partially required for inducing PHO genes. Phylogenetic survey suggests that reduced dependence on Pho2 evolved in C. glabrata and closely related species. In S. cerevisiae, less Pho2-dependent Pho4 orthologs induce more genes. In C. glabrata, its Pho4 binds to more locations and induces three times as many genes as Pho4 in S. cerevisiae does. Our work shows how evolution of combinatorial regulation allows for rapid expansion of a gene regulatory network's targets, possibly extending its physiological functions.
Keywords: C. glabrata; Hemiascomycetes; S. cerevisiae; evolution of gene regulatory network; evolutionary biology; genomics; infectious disease; microbiology; other Hemiascomycete yeast species; phosphate starvation response.
Plain language summary
The diversity of life on Earth has intrigued generations of scientists and nature lovers alike. Research over recent decades has revealed that much of the diversity we can see did not require the invention of new genes. Instead, living forms diversified mostly by using old genes in new ways – for example, by changing when or where an existing gene became active. This kind of change is referred to as “regulatory evolution”. A class of proteins called transcription factors are hot spots in regulatory evolution. These proteins recognize specific sequences of DNA to control the activity of other genes, and so represent the “readers” of the genetic information. Small changes to how a transcription factor is regulated, or the genes it targets, can lead to dramatic changes in an organism. Before we can understand how life on Earth evolved to be so diverse, scientists must first answer how transcription factors evolve and what consequences this has on their target genes. So far, most studies of regulatory evolution have focused on networks of transcription factors and genes that control how an organism develops. He et al. have now studied a regulatory network that is behind a different process, namely how an organism responds to stress or starvation. These two types of regulatory networks are structured differently and work in different ways. These differences made He et al. wonder if the networks evolved differently too. The chemical phosphate is an essential nutrient for all living things, and He et al. compared how two different species of yeast responded to a lack of phosphate. The key difference was how much a major transcription factor known as Pho4 depended on a so-called co-activator protein named Pho2 to carry out its role. Baker’s yeast (Saccharomyces cerevisiae), which is commonly used in laboratory experiments, requires both Pho4 and Pho2 to activate about 20 genes when inorganic phosphate is not available in its environment. However, in a related yeast species called Candida glabrata, Pho4 has evolved to depend less on Pho2. He et al. went on to show that, as well as being less dependent on Pho2, Pho4 in C. glabrata activates more than three times as many genes as Pho4 in S. cerevisiae does in the absence of phosphate. These additional gene targets for Pho4 in C. glabrata are predicted to extend the network’s activities, and allow it to regulate new process including the yeast’s responses to other types of stress and the building of the yeast’s cell wall. Together these findings show a new way that regulatory networks can evolve, that is, by reducing its dependence on the co-activator, a transcription factor can expand the number of genes it targets. This has not been seen for regulatory networks related to development, suggesting that different networks can indeed evolve in different ways. Lastly, because disease-causing microbes are often stressed inside their hosts and C. glabrata sometimes infects humans, understanding how this yeast’s response to stress has evolved may lead to new ways to prevent and treat this infection.
Conflict of interest statement
No competing interests declared.
Chief Scientific Officer and a Vice President at the Howard Hughes Medical Institute, one of the three founding funders of eLife.
Figures
References
-
- Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, Grüning B, Guerler A, Hillman-Jackson J, Von Kuster G, Rasche E, Soranzo N, Turaga N, Taylor J, Nekrutenko A, Goecks J. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Research. 2016;44:W3–W10. doi: 10.1093/nar/gkw343. - DOI - PMC - PubMed
-
- Atanasova R, Angoulvant A, Tefit M, Gay F, Guitard J, Mazier D, Fairhead C, Hennequin C. A mouse model for Candida glabrata hematogenous disseminated infection starting from the gut: evaluation of strains with different adhesion properties. PLoS One. 2013;8:e69664. doi: 10.1371/journal.pone.0069664. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
