

How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy
- PMID: 28485722
- PMCID: PMC5623086
- DOI: 10.1038/gt.2017.34
How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy
Abstract
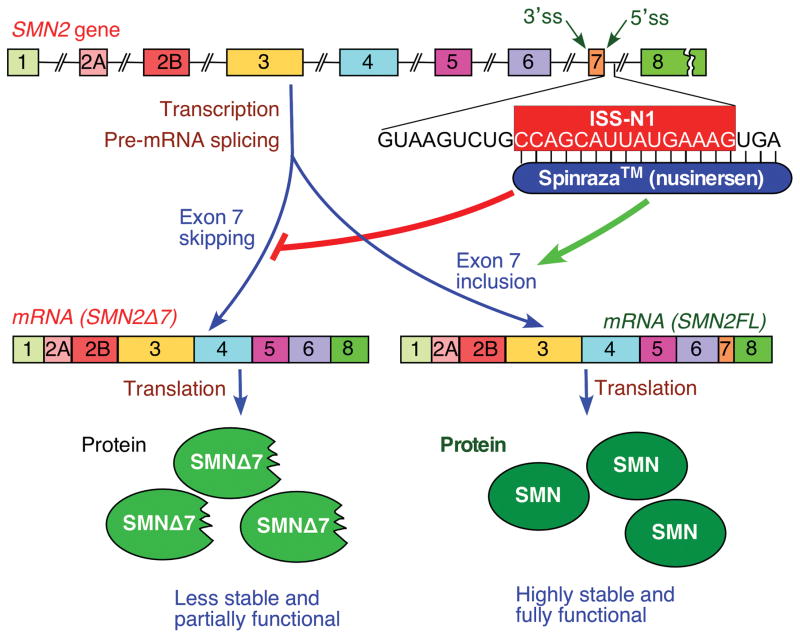
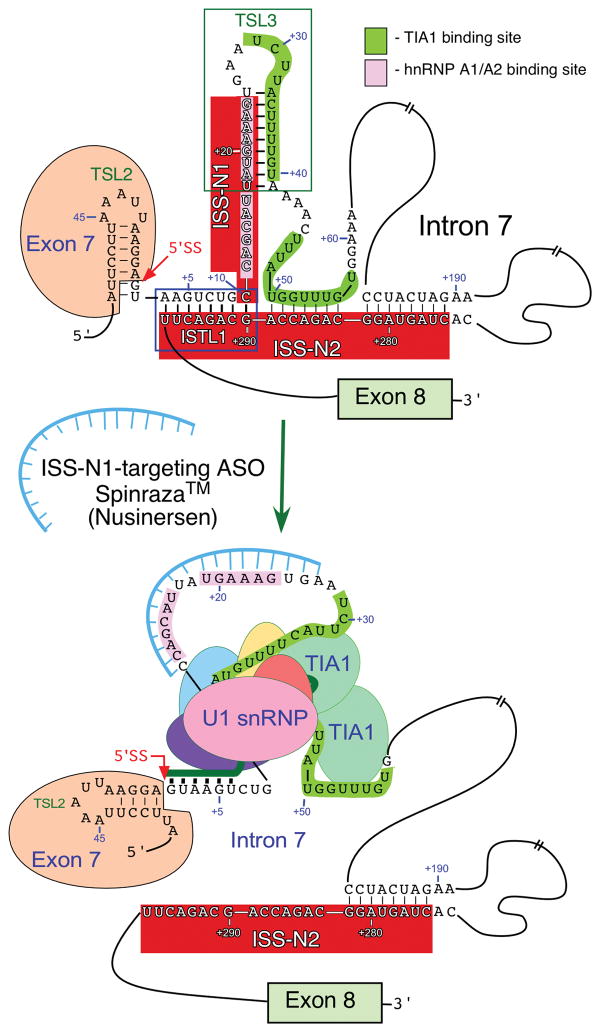
Spinal muscular atrophy (SMA), a prominent genetic disease of infant mortality, is caused by low levels of survival motor neuron (SMN) protein owing to deletions or mutations of the SMN1 gene. SMN2, a nearly identical copy of SMN1 present in humans, cannot compensate for the loss of SMN1 because of predominant skipping of exon 7 during pre-mRNA splicing. With the recent US Food and Drug Administration approval of nusinersen (Spinraza), the potential for correction of SMN2 exon 7 splicing as an SMA therapy has been affirmed. Nusinersen is an antisense oligonucleotide that targets intronic splicing silencer N1 (ISS-N1) discovered in 2004 at the University of Massachusetts Medical School. ISS-N1 has emerged as the model target for testing the therapeutic efficacy of antisense oligonucleotides using different chemistries as well as different mouse models of SMA. Here, we provide a historical account of events that led to the discovery of ISS-N1 and describe the impact of independent validations that raised the profile of ISS-N1 as one of the most potent antisense targets for the treatment of a genetic disease. Recent approval of nusinersen provides a much-needed boost for antisense technology that is just beginning to realize its potential. Beyond treating SMA, the ISS-N1 target offers myriad potentials for perfecting various aspects of the nucleic-acid-based technology for the amelioration of the countless number of pathological conditions.
Figures
References
-
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. - PubMed
-
- Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) Hum Mutat. 2000;15:228–237. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
