

The amyloid-β oligomer Aβ*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation
- PMID: 28487416
- PMCID: PMC5859319
- DOI: 10.1126/scisignal.aal2021
The amyloid-β oligomer Aβ*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation
Retraction in
-
Retraction.Sci Signal. 2025 Dec 23;18(918):eaee6619. doi: 10.1126/scisignal.aee6619. Epub 2025 Dec 23. Sci Signal. 2025. PMID: 41433422 No abstract available.
Expression of concern in
-
Editorial expression of concern.Sci Signal. 2022 Jun 21;15(739):eadd4322. doi: 10.1126/scisignal.add4322. Epub 2022 Jun 21. Sci Signal. 2022. PMID: 35727863 No abstract available.
Abstract
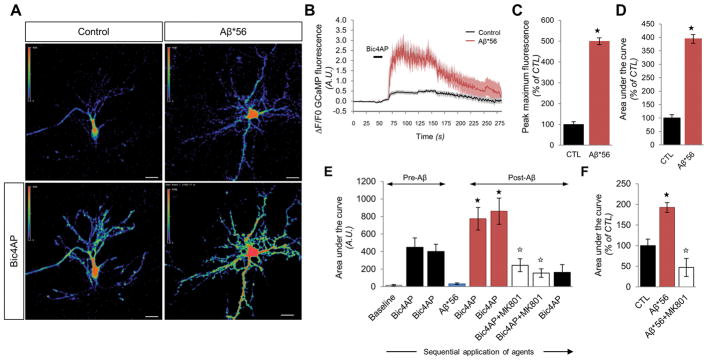
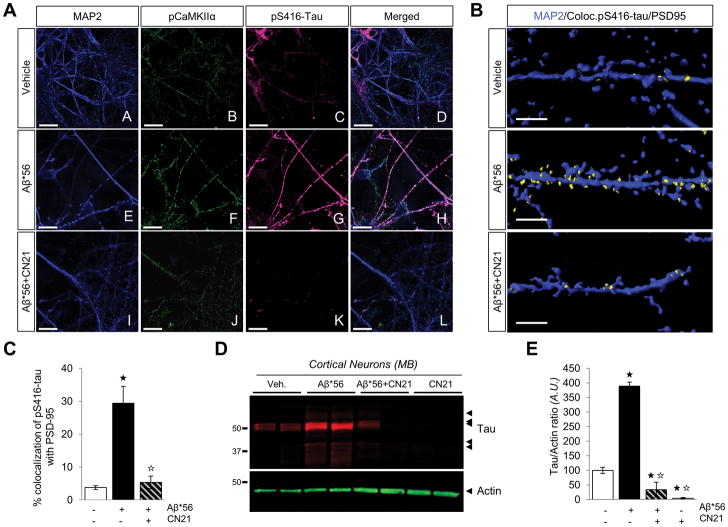
Oligomeric forms of amyloid-forming proteins are believed to be the principal initiating bioactive species in many neurodegenerative disorders, including Alzheimer's disease (AD). Amyloid-β (Aβ) oligomers are implicated in AD-associated phosphorylation and aggregation of the microtubule-associated protein tau. To investigate the specific molecular pathways activated by different assemblies, we isolated various forms of Aβ from Tg2576 mice, which are a model for AD. We found that Aβ*56, a 56-kDa oligomer that is detected before patients develop overt signs of AD, induced specific changes in neuronal signaling. In primary cortical neurons, Aβ*56 interacted with N-methyl-d-aspartate receptors (NMDARs), increased NMDAR-dependent Ca2+ influx, and consequently increased intracellular calcium concentrations and the activation of Ca2+-dependent calmodulin kinase IIα (CaMKIIα). In cultured neurons and in the brains of Tg2576 mice, activated CaMKIIα was associated with increased site-specific phosphorylation and missorting of tau, both of which are associated with AD pathology. In contrast, exposure of cultured primary cortical neurons to other oligomeric Aβ forms (dimers and trimers) did not trigger these effects. Our results indicate that distinct Aβ assemblies activate neuronal signaling pathways in a selective manner and that dissecting the molecular events caused by each oligomer may inform more effective therapeutic strategies.
Copyright © 2017, American Association for the Advancement of Science.
Conflict of interest statement
Figures





References
-
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. - PubMed
-
- Ittner LM, Gotz J. Amyloid-beta and tau--a toxic pas de deux in Alzheimer’s disease. Nature reviews Neuroscience. 2011;12:65–72. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
