

Common sequence variants affect molecular function more than rare variants?
- PMID: 28487536
- PMCID: PMC5431670
- DOI: 10.1038/s41598-017-01054-2
Common sequence variants affect molecular function more than rare variants?
Abstract
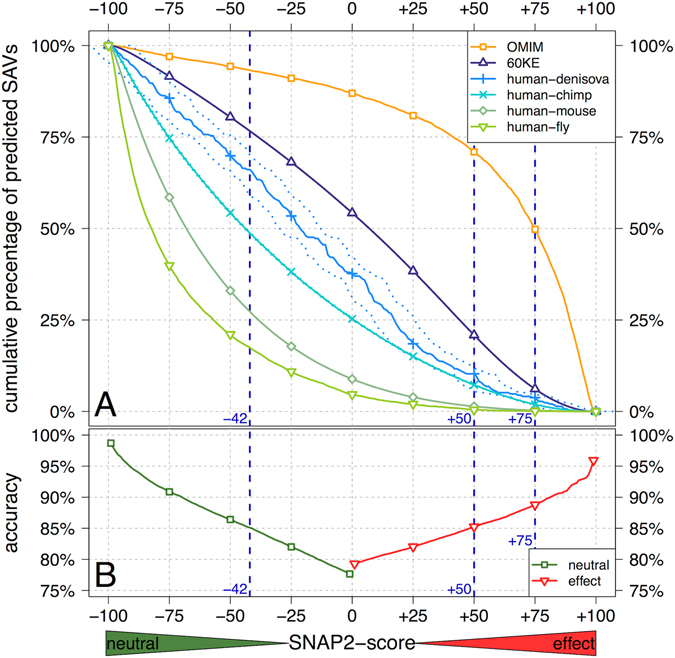
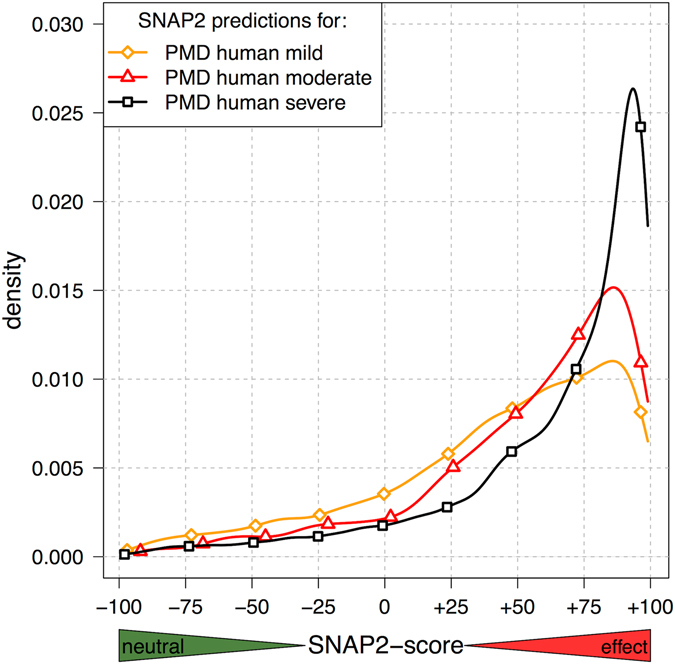
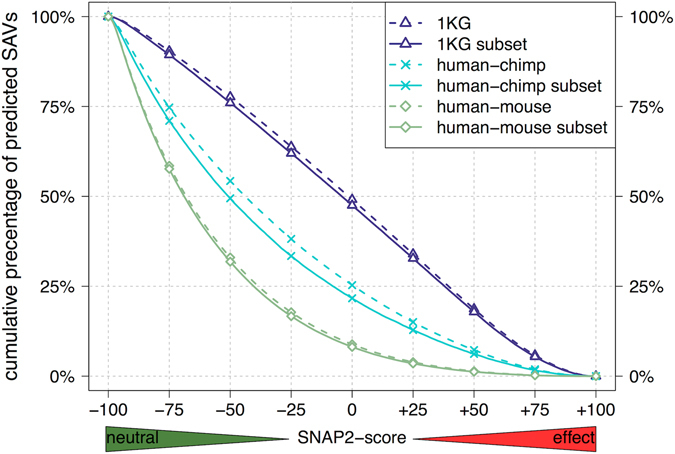
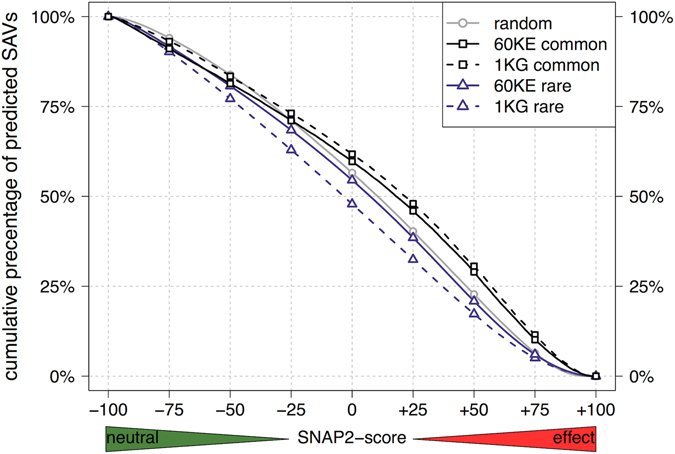
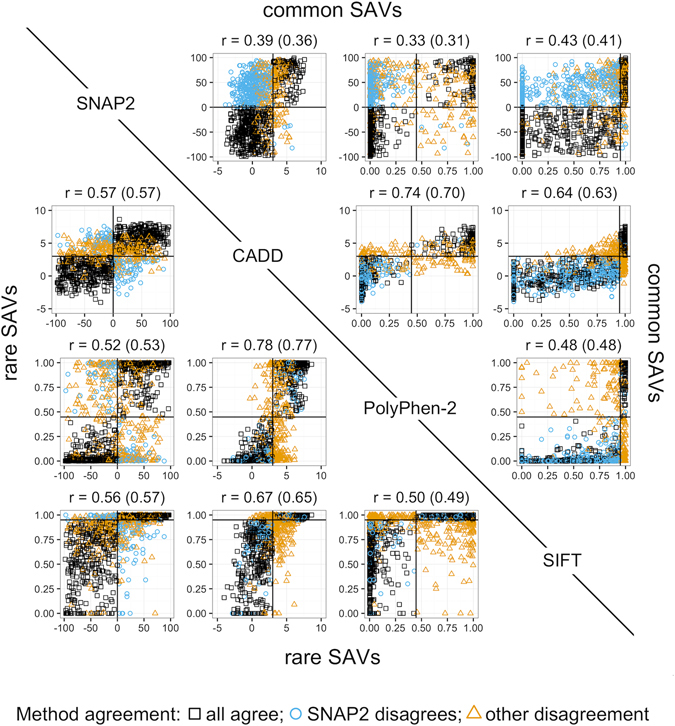
Any two unrelated individuals differ by about 10,000 single amino acid variants (SAVs). Do these impact molecular function? Experimental answers cannot answer comprehensively, while state-of-the-art prediction methods can. We predicted the functional impacts of SAVs within human and for variants between human and other species. Several surprising results stood out. Firstly, four methods (CADD, PolyPhen-2, SIFT, and SNAP2) agreed within 10 percentage points on the percentage of rare SAVs predicted with effect. However, they differed substantially for the common SAVs: SNAP2 predicted, on average, more effect for common than for rare SAVs. Given the large ExAC data sets sampling 60,706 individuals, the differences were extremely significant (p-value < 2.2e-16). We provided evidence that SNAP2 might be closer to reality for common SAVs than the other methods, due to its different focus in development. Secondly, we predicted significantly higher fractions of SAVs with effect between healthy individuals than between species; the difference increased for more distantly related species. The same trends were maintained for subsets of only housekeeping proteins and when moving from exomes of 1,000 to 60,000 individuals. SAVs frozen at speciation might maintain protein function, while many variants within a species might bring about crucial changes, for better or worse.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
