

Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration
- PMID: 28489072
- PMCID: PMC5436137
- DOI: 10.1038/ncomms15090
Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration
Abstract
Oncogenic Ras mutants, frequently detected in human cancers, are high-priority anticancer drug targets. However, direct inhibition of oncogenic Ras mutants with small molecules has been extremely challenging. Here we report the development of a human IgG1 format antibody, RT11, which internalizes into the cytosol of living cells and selectively binds to the activated GTP-bound form of various oncogenic Ras mutants to block the interactions with effector proteins, thereby suppressing downstream signalling and exerting anti-proliferative effects in a variety of tumour cells harbouring oncogenic Ras mutants. When systemically administered, an RT11 variant with an additional tumour-associated integrin binding moiety for tumour tissue targeting significantly inhibits the in vivo growth of oncogenic Ras-mutated tumour xenografts in mice, but not wild-type Ras-harbouring tumours. Our results demonstrate the feasibility of developing therapeutic antibodies for direct targeting of cytosolic proteins that are inaccessible using current antibody technology.
Conflict of interest statement
S.M.S., D.K.C. and Y.S.K. are listed as inventors on pending patent applications (PCT/KR2015/007626 and PCT/KR2015/007627) related to technology described in this work. These remaining authors declare no competing financial interests.
Figures
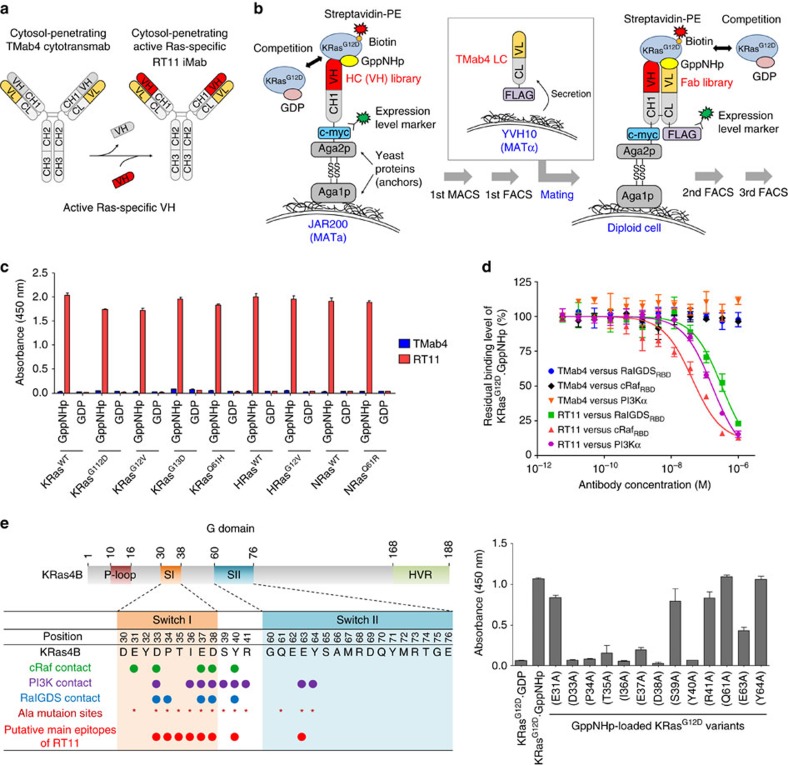
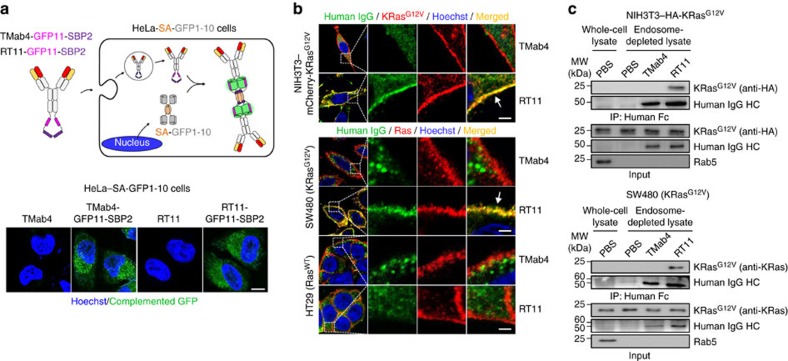
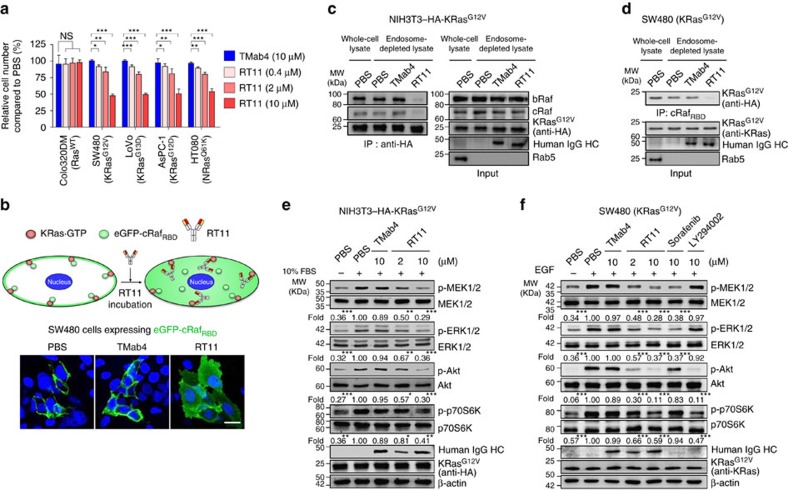
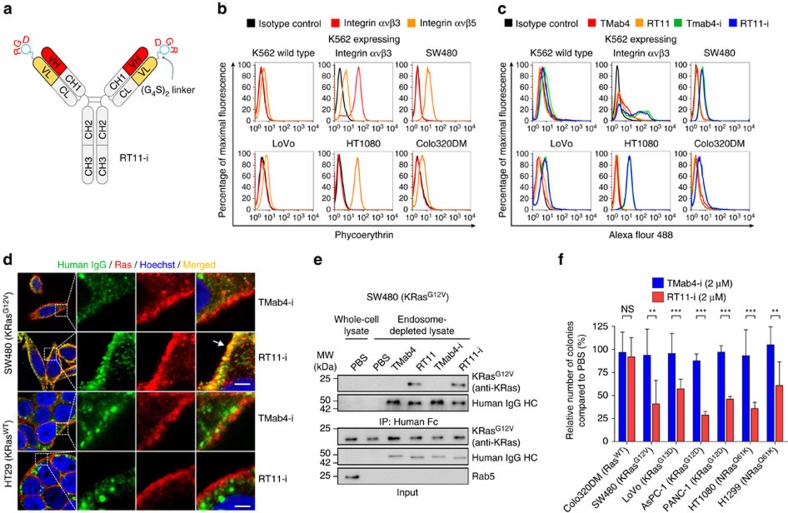
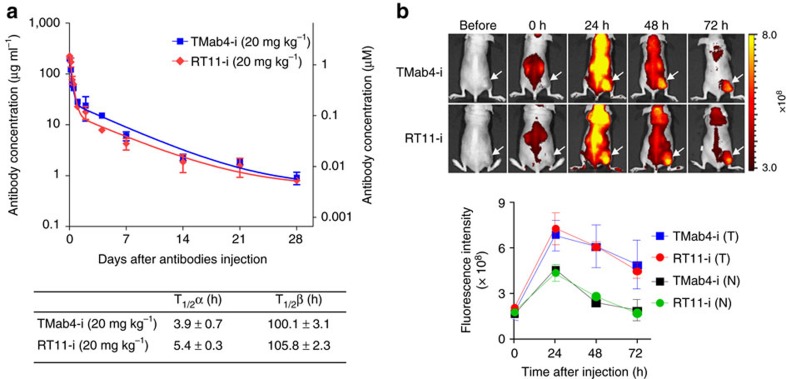
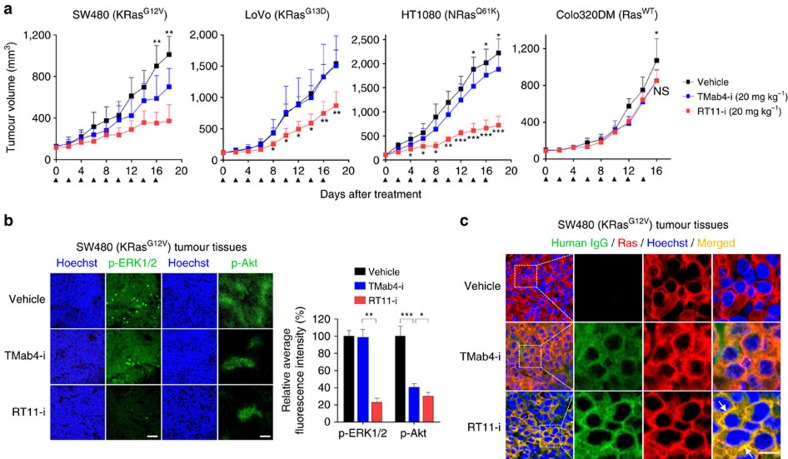
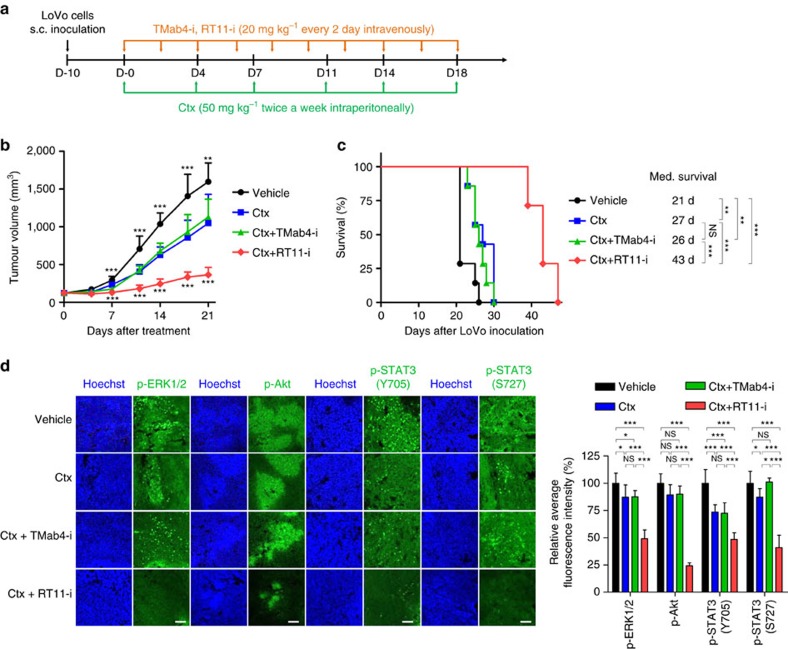
References
-
- Stephen A. G., Esposito D., Bagni R. K. & McCormick F. Dragging ras back in the ring. Cancer Cell 25, 272–281 (2014). - PubMed
-
- Milburn M. V. et al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science 247, 939–945 (1990). - PubMed
-
- Vetter I. R. & Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304 (2001). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
