

AKT Inhibition in Solid Tumors With AKT1 Mutations
- PMID: 28489509
- PMCID: PMC5501365
- DOI: 10.1200/JCO.2017.73.0143
AKT Inhibition in Solid Tumors With AKT1 Mutations
Erratum in
-
Errata.J Clin Oncol. 2019 Feb 1;37(4):360. doi: 10.1200/JCO.18.02209. J Clin Oncol. 2019. PMID: 30695659 Free PMC article. No abstract available.
Abstract
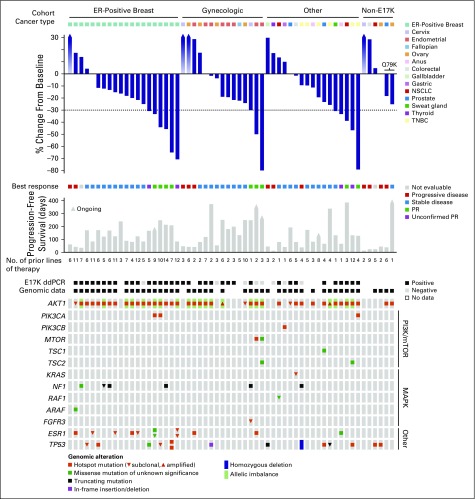
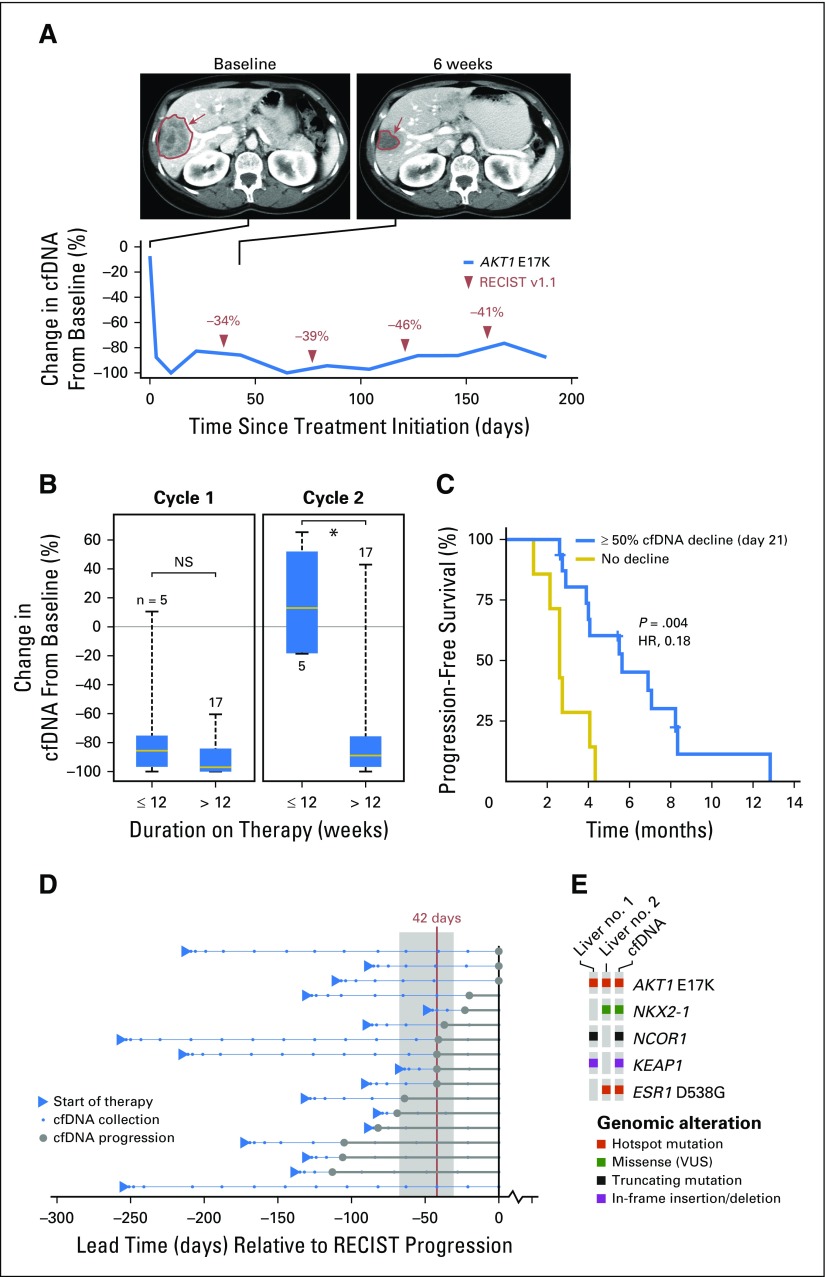
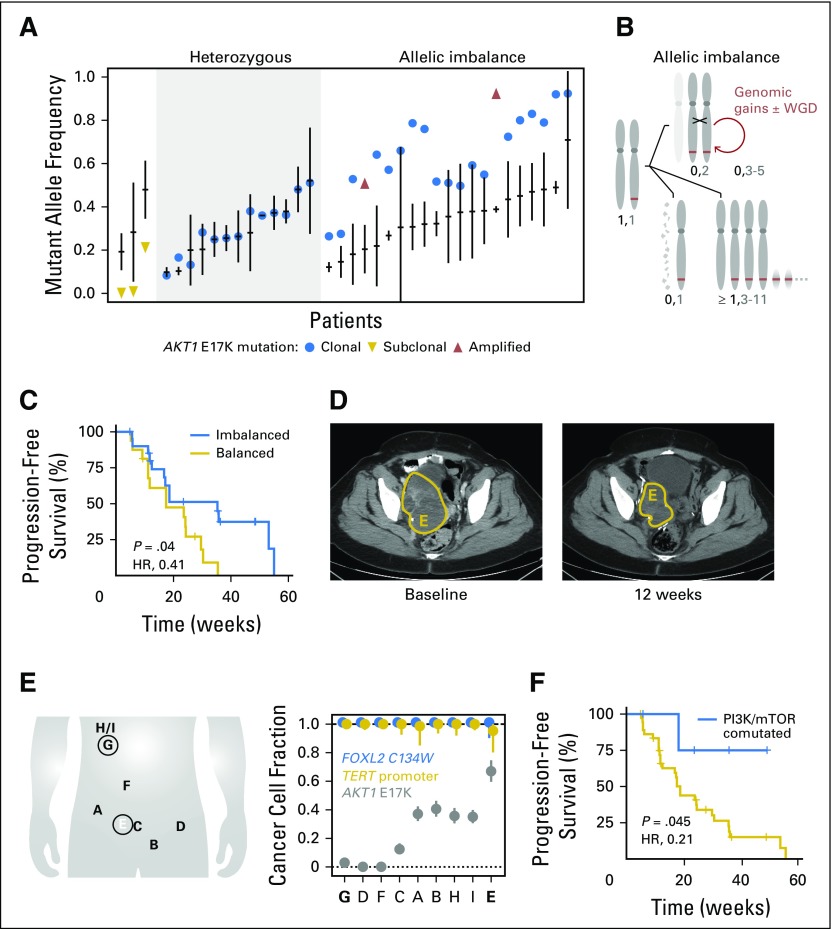
Purpose AKT1 E17K mutations are oncogenic and occur in many cancers at a low prevalence. We performed a multihistology basket study of AZD5363, an ATP-competitive pan-AKT kinase inhibitor, to determine the preliminary activity of AKT inhibition in AKT-mutant cancers. Patients and Methods Fifty-eight patients with advanced solid tumors were treated. The primary end point was safety; secondary end points were progression-free survival (PFS) and response according to Response Evaluation Criteria in Solid Tumors (RECIST). Tumor biopsies and plasma cell-free DNA (cfDNA) were collected in the majority of patients to identify predictive biomarkers of response. Results In patients with AKT1 E17K-mutant tumors (n = 52) and a median of five lines of prior therapy, the median PFS was 5.5 months (95% CI, 2.9 to 6.9 months), 6.6 months (95% CI, 1.5 to 8.3 months), and 4.2 months (95% CI, 2.1 to 12.8 months) in patients with estrogen receptor-positive breast, gynecologic, and other solid tumors, respectively. In an exploratory biomarker analysis, imbalance of the AKT1 E17K-mutant allele, most frequently caused by copy-neutral loss-of-heterozygosity targeting the wild-type allele, was associated with longer PFS (hazard ratio [HR], 0.41; P = .04), as was the presence of coincident PI3K pathway hotspot mutations (HR, 0.21; P = .045). Persistent declines in AKT1 E17K in cfDNA were associated with improved PFS (HR, 0.18; P = .004) and response ( P = .025). Responses were not restricted to patients with detectable AKT1 E17K in pretreatment cfDNA. The most common grade ≥ 3 adverse events were hyperglycemia (24%), diarrhea (17%), and rash (15.5%). Conclusion This study provides the first clinical data that AKT1 E17K is a therapeutic target in human cancer. The genomic context of the AKT1 E17K mutation further conditioned response to AZD5363.
Figures
References
-
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. - PubMed
-
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. - PubMed
-
- Engelman JA. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. - PubMed
-
- Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. - PubMed
-
- Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–2963. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
