

Metabolic Reprogramming in Glioma
- PMID: 28491867
- PMCID: PMC5405080
- DOI: 10.3389/fcell.2017.00043
Metabolic Reprogramming in Glioma
Abstract
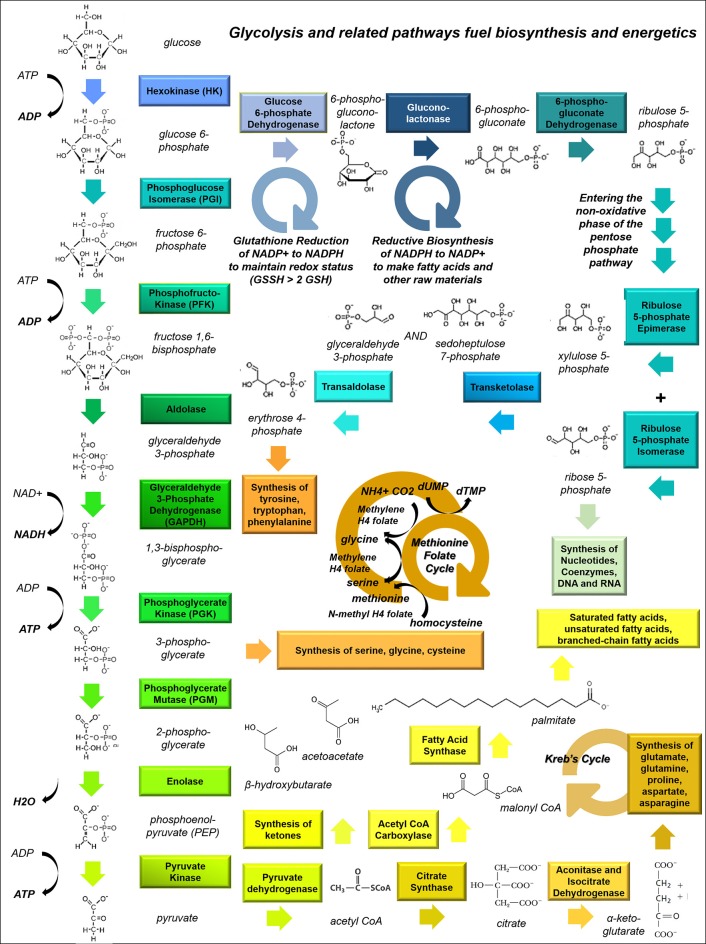
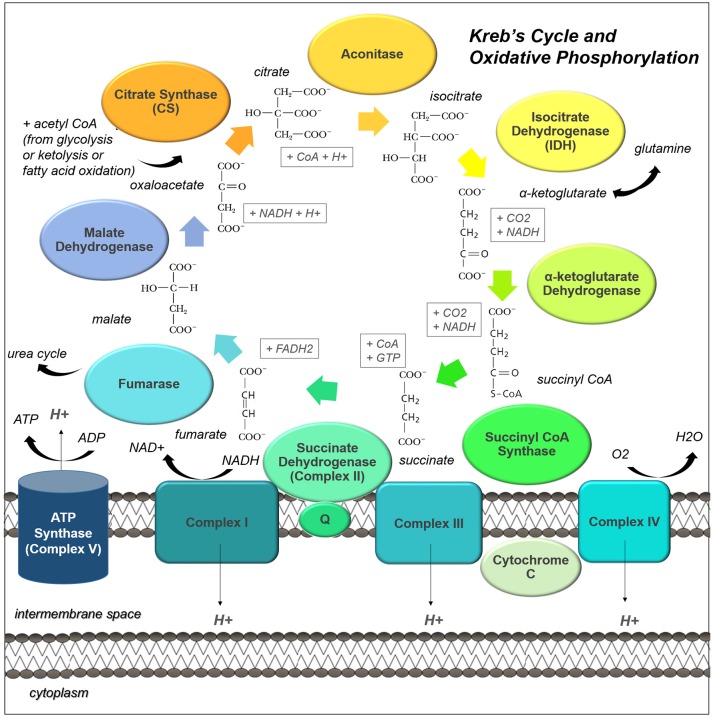
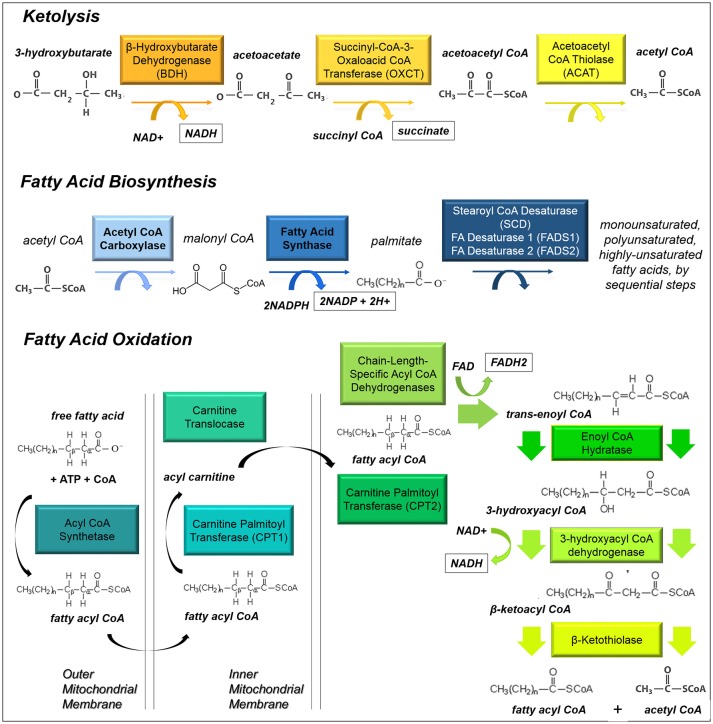
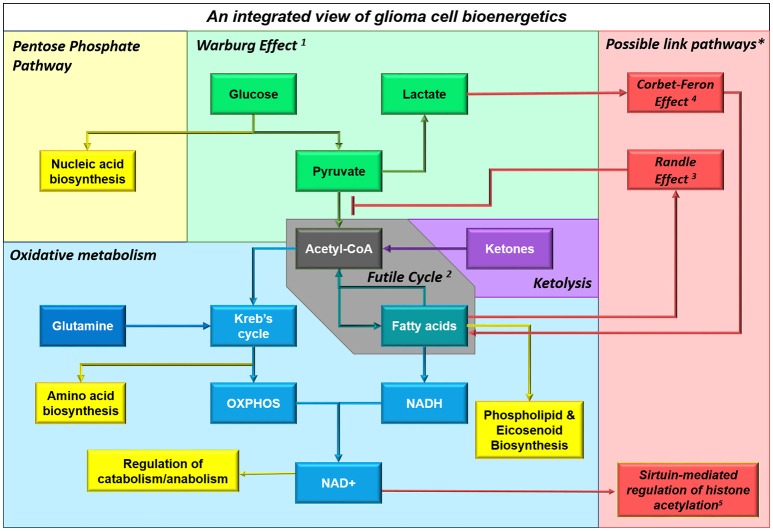
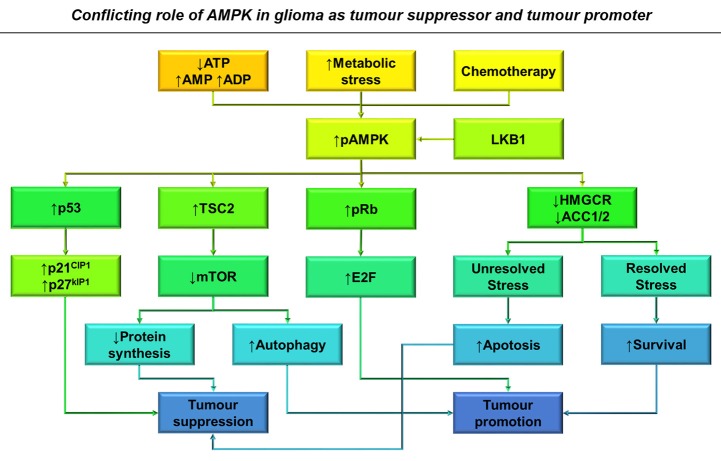
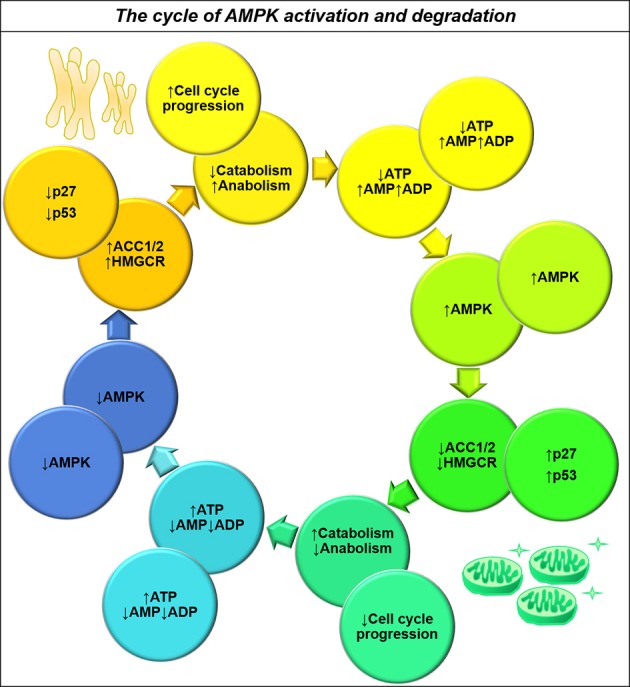
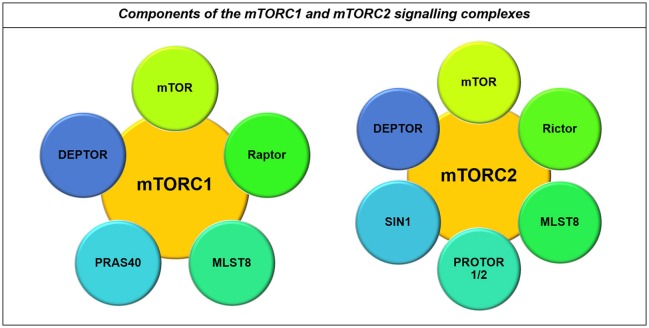
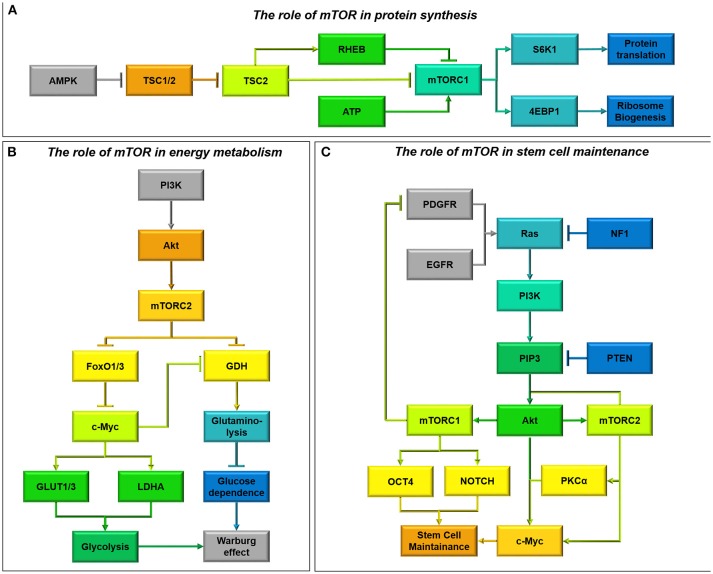
Many cancers have long been thought to primarily metabolize glucose for energy production-a phenomenon known as the Warburg Effect, after the classic studies of Otto Warburg in the early twentieth century. Yet cancer cells also utilize other substrates, such as amino acids and fatty acids, to produce raw materials for cellular maintenance and energetic currency to accomplish cellular tasks. The contribution of these substrates is increasingly appreciated in the context of glioma, the most common form of malignant brain tumor. Multiple catabolic pathways are used for energy production within glioma cells, and are linked in many ways to anabolic pathways supporting cellular function. For example: glycolysis both supports energy production and provides carbon skeletons for the synthesis of nucleic acids; meanwhile fatty acids are used both as energetic substrates and as raw materials for lipid membranes. Furthermore, bio-energetic pathways are connected to pro-oncogenic signaling within glioma cells. For example: AMPK signaling links catabolism with cell cycle progression; mTOR signaling contributes to metabolic flexibility and cancer cell survival; the electron transport chain produces ATP and reactive oxygen species (ROS) which act as signaling molecules; Hypoxia Inducible Factors (HIFs) mediate interactions with cells and vasculature within the tumor environment. Mutations in the tumor suppressor p53, and the tricarboxylic acid cycle enzymes Isocitrate Dehydrogenase 1 and 2 have been implicated in oncogenic signaling as well as establishing metabolic phenotypes in genetically-defined subsets of malignant glioma. These pathways critically contribute to tumor biology. The aim of this review is two-fold. Firstly, we present the current state of knowledge regarding the metabolic strategies employed by malignant glioma cells, including aerobic glycolysis; the pentose phosphate pathway; one-carbon metabolism; the tricarboxylic acid cycle, which is central to amino acid metabolism; oxidative phosphorylation; and fatty acid metabolism, which significantly contributes to energy production in glioma cells. Secondly, we highlight processes (including the Randle Effect, AMPK signaling, mTOR activation, etc.) which are understood to link bio-energetic pathways with oncogenic signals, thereby allowing the glioma cell to achieve a pro-malignant state.
Keywords: autophagy; biosynthesis; brain tumors; cancer; catabolism; glioma; metabolism; mitochondria.
Figures
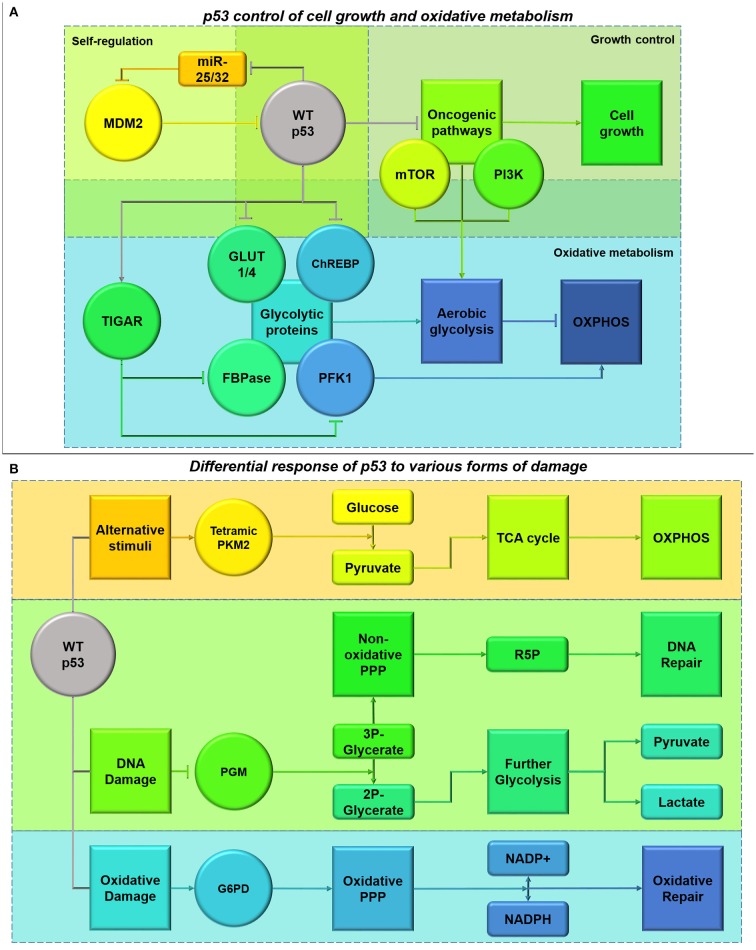
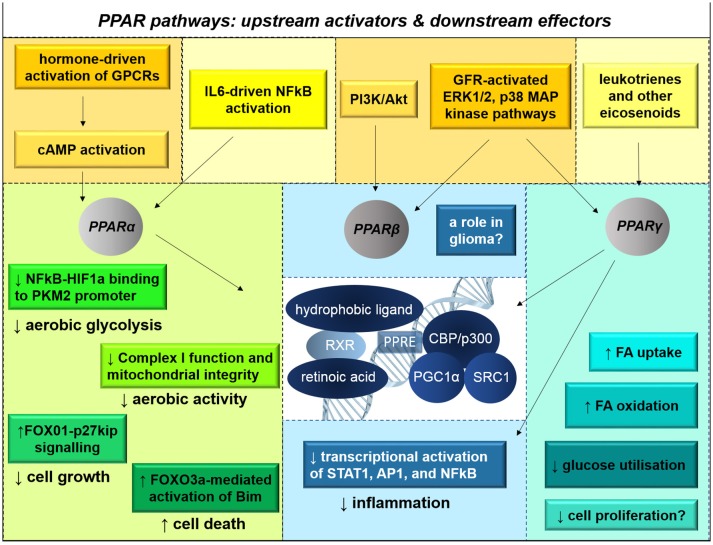
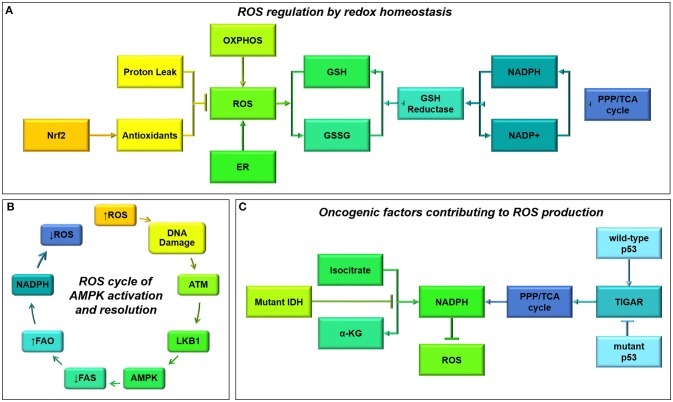
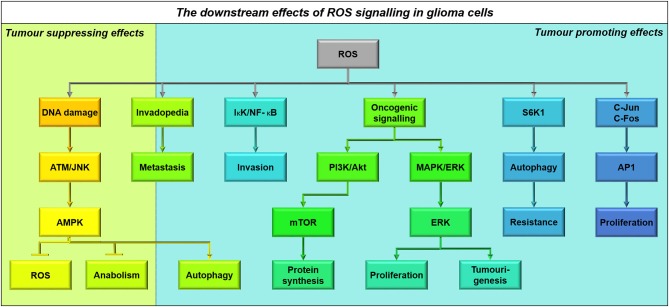
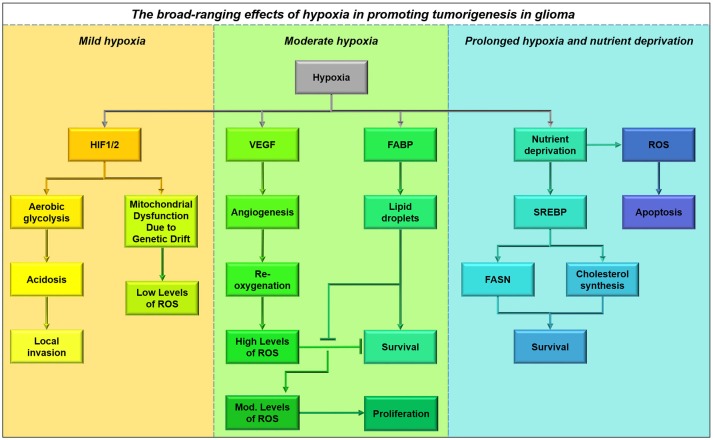
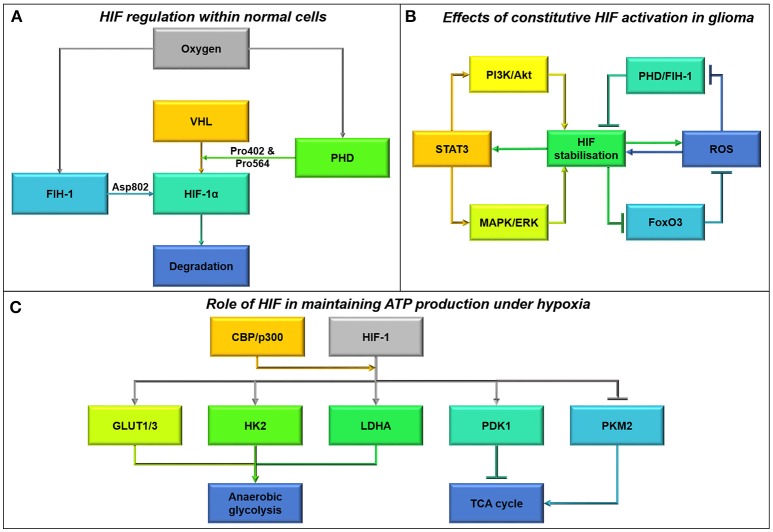
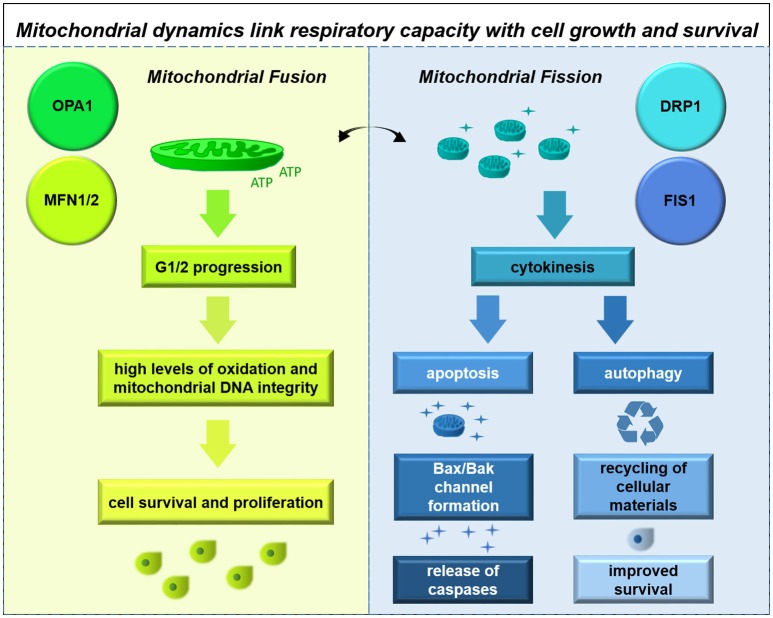
References
-
- Albert N. L., Weller M., Suchorska B., Galldiks N., Soffietti R., Kim M. M., et al. . (2016). Response assessment in neuro-oncology working group and european association for neuro-oncology recommendations for the clinical use of PET imaging in gliomas. Neuro-oncology 18, 1199–1208. 10.1093/neuonc/now058 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous