

Circulating tumour DNA sequence analysis as an alternative to multiple myeloma bone marrow aspirates
- PMID: 28492226
- PMCID: PMC5437268
- DOI: 10.1038/ncomms15086
Circulating tumour DNA sequence analysis as an alternative to multiple myeloma bone marrow aspirates
Abstract
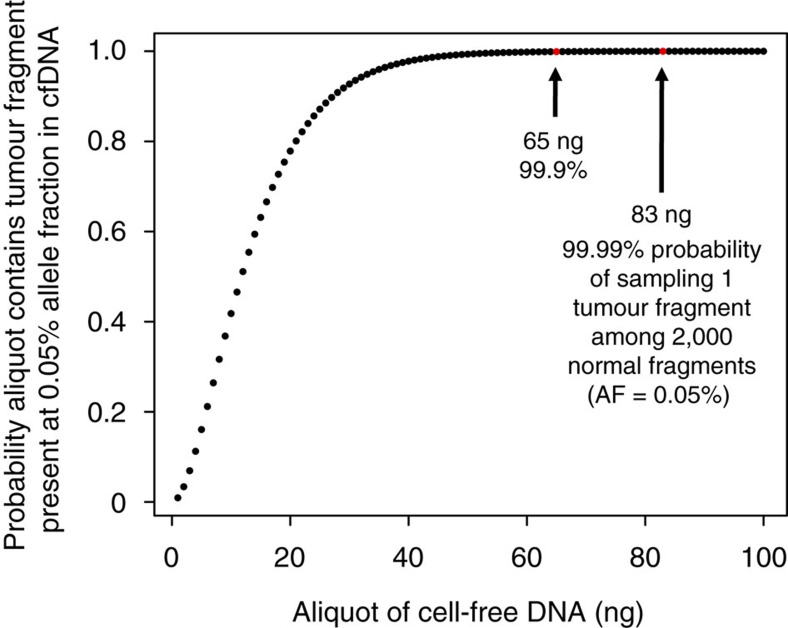
The requirement for bone-marrow aspirates for genomic profiling of multiple myeloma poses an obstacle to enrolment and retention of patients in clinical trials. We evaluated whether circulating cell-free DNA (cfDNA) analysis is comparable to molecular profiling of myeloma using bone-marrow tumour cells. We report here a hybrid-capture-based Liquid Biopsy Sequencing (LB-Seq) method used to sequence all protein-coding exons of KRAS, NRAS, BRAF, EGFR and PIK3CA in 64 cfDNA specimens from 53 myeloma patients to >20,000 × median coverage. This method includes a variant filtering algorithm that enables detection of tumour-derived fragments present in cfDNA at allele frequencies as low as 0.25% (median 3.2%, range 0.25-46%). Using LB-Seq analysis of 48 cfDNA specimens with matched bone-marrow data, we detect 49/51 likely somatic mutations, with subclonal hierarchies reflecting tumour profiling (96% concordance), and four additional mutations likely missed by bone-marrow testing (>98% specificity). Overall, LB-Seq is a high fidelity adjunct to genetic profiling of bone-marrow in multiple myeloma.
Conflict of interest statement
S.T. received research support from GlaxoSmithKline, Amgen, Oncoethix and Astellas and provided consultant services to Novartis. T.J.P. received research support from Boehringer Ingelheim. The remaining authors declare no competing financial interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
