

Pemphigus
- PMID: 28492232
- PMCID: PMC5901732
- DOI: 10.1038/nrdp.2017.26
Pemphigus
Abstract
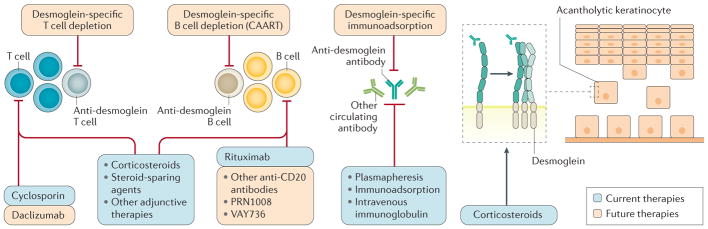
Pemphigus is a group of IgG-mediated autoimmune diseases of stratified squamous epithelia, such as the skin and oral mucosa, in which acantholysis (the loss of cell adhesion) causes blisters and erosions. Pemphigus has three major subtypes: pemphigus vulgaris, pemphigus foliaceus and paraneoplastic pemphigus. IgG autoantibodies are characteristically raised against desmoglein 1 and desmoglein 3, which are cell-cell adhesion molecules found in desmosomes. The sites of blister formation can be physiologically explained by the anti-desmoglein autoantibody profile and tissue-specific expression pattern of desmoglein isoforms. The pathophysiological roles of T cells and B cells have been characterized in mouse models of pemphigus and patients, revealing insights into the mechanisms of autoimmunity. Diagnosis is based on clinical manifestations and confirmed with histological and immunochemical testing. The current first-line treatment is systemic corticosteroids and adjuvant therapies, including immunosuppressive agents, intravenous immunoglobulin and plasmapheresis. Rituximab, a monoclonal antibody against CD20+ B cells, is a promising therapeutic option that may soon become first-line therapy. Pemphigus is one of the best-characterized human autoimmune diseases and provides an ideal paradigm for both basic and clinical research, especially towards the development of antigen-specific immune suppression treatments for autoimmune diseases.
Conflict of interest statement
M.A. receives speaker honoraria and grants from Nihon Pharmaceutical, research support from Medical & Biological Laboratories, Health Sciences Research Grants for Research on Rare and Intractable Diseases from Ministry of Health, Labour, and Welfare, and grants from Japan Society for the Promotion of Science and Agency for Medical Research and Development. H.T. receives grants from Japan Society for the Promotion of Science. A.S.P. is a consultant for Syntimmune and TG Therapeutics and receives grants or research support from the US NIH (R01-AR057001, R56-AR064220 and R01-068288), the Dermatology Foundation and Sanofi. She is co-inventor on a patent related to chimeric immunoreceptor therapy of pemphigus. The content is solely the responsibility of the authors and does not necessarily represent the official views of the US NIH. C.T.E. receives grants or research support from Deutsche Forschungsgemeinschaft (EL711/1-1) and is co-inventor on a patent related to chimeric immunoreceptor therapy of pemphigus. D.Z. is an advisory board member for Roche Pharma, and a consultant for Euroimmun, Almirall, UCB, Fresenius and arGEN-X. He received speakers honoraria and/or travel/accommodations/meeting compensation from Biotest, Fresenius, Miltenyi, Roche Pharma, Biogen Idec, AbbVie, UCB, Janssen and grants or research support from Euroimmun, Miltenyi, Fresenius, Biotest, Dompe, Almirall, Biogen and Roche. He holds the patent Euroimmun: DE 10 2006 059 574 A1. M.K. and J.Y. declare no competing interests.
Figures






Comment in
-
Authors' reply: Paraneoplastic autoimmune multi-organ syndrome is a distinct entity from traditional pemphigus subtypes.Nat Rev Dis Primers. 2018 Feb 22;4:18013. doi: 10.1038/nrdp.2018.13. Nat Rev Dis Primers. 2018. PMID: 29469088 No abstract available.
-
Paraneoplastic autoimmune multi-organ syndrome is a distinct entity from traditional pemphigus subtypes.Nat Rev Dis Primers. 2018 Feb 22;4:18012. doi: 10.1038/nrdp.2018.12. Nat Rev Dis Primers. 2018. PMID: 29469089 No abstract available.
References
-
- Amagai M. In: Dermatology. Bolognia JL, Jorizzo JL, Schaffer JV, editors. Vol. 1. Mosby; 2012. pp. 461–474. Ch. 29.
-
- Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67:869–877. This study reports the cDNA cloning of a pemphigus vulgaris antigen, which turned out to be a desmosomal cadherin called desmoglein 3. - PubMed
-
- Koch PJ, et al. Identification of desmoglein, a constitutive desmosomal glycoprotein, as a member of the cadherin family of cell adhesion molecules. Eur J Cell Biol. 1990;53:1–12. - PubMed
-
- Stanley JR, Amagai M. Pemphigus, bullous impetigo, and the staphylococcal scalded-skin syndrome. N Engl J Med. 2006;355:1800–1810. - PubMed
-
- Anhalt GJ, et al. Paraneoplastic pemphigus. An autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med. 1990;323:1729–1735. - PubMed
