

Local genes for local bacteria: Evidence of allopatry in the genomes of transatlantic Campylobacter populations
- PMID: 28493321
- PMCID: PMC5600125
- DOI: 10.1111/mec.14176
Local genes for local bacteria: Evidence of allopatry in the genomes of transatlantic Campylobacter populations
Abstract
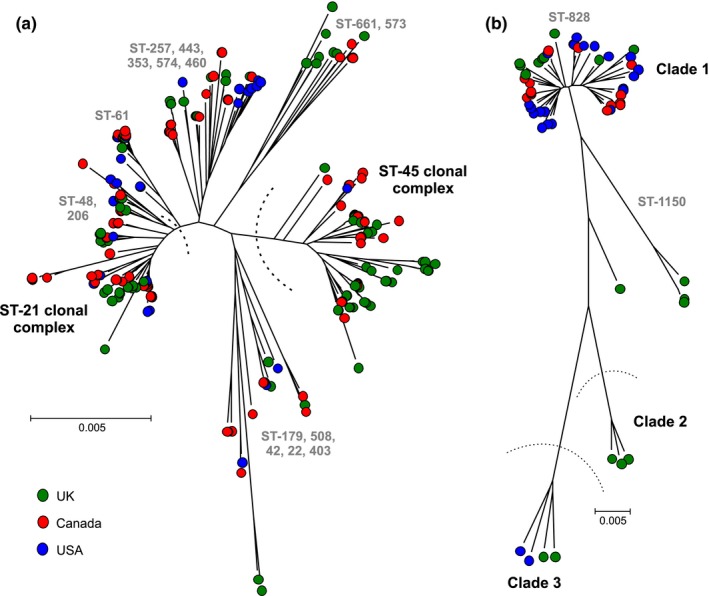
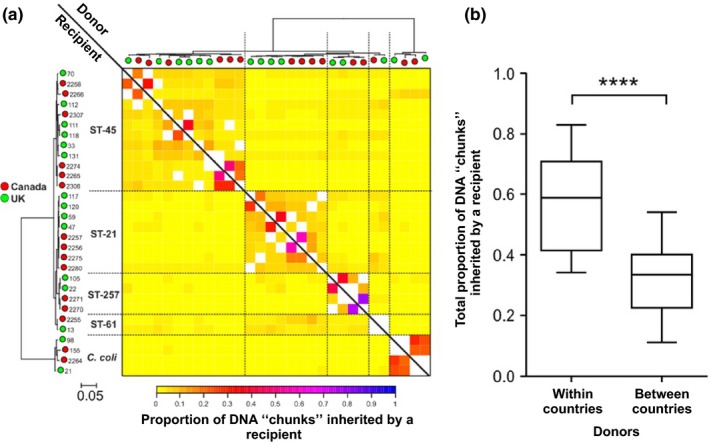
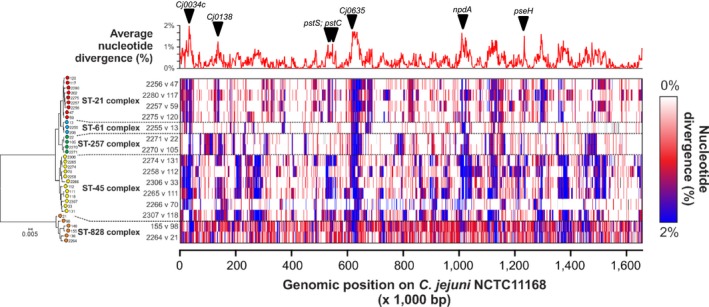
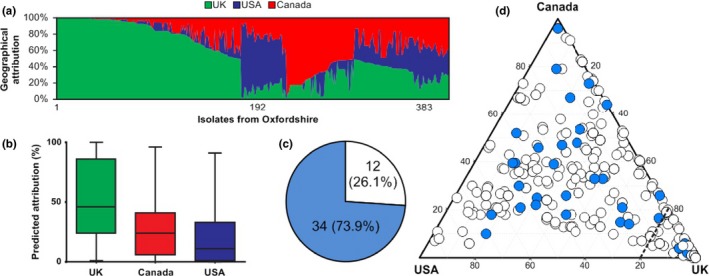
The genetic structure of bacterial populations can be related to geographical locations of isolation. In some species, there is a strong correlation between geographical distance and genetic distance, which can be caused by different evolutionary mechanisms. Patterns of ancient admixture in Helicobacter pylori can be reconstructed in concordance with past human migration, whereas in Mycobacterium tuberculosis it is the lack of recombination that causes allopatric clusters. In Campylobacter, analyses of genomic data and molecular typing have been successful in determining the reservoir host species, but not geographical origin. We investigated biogeographical variation in highly recombining genes to determine the extent of clustering between genomes from geographically distinct Campylobacter populations. Whole-genome sequences from 294 Campylobacter isolates from North America and the UK were analysed. Isolates from within the same country shared more recently recombined DNA than isolates from different countries. Using 15 UK/American closely matched pairs of isolates that shared ancestors, we identify regions that have frequently and recently recombined to test their correlation with geographical origin. The seven genes that demonstrated the greatest clustering by geography were used in an attribution model to infer geographical origin which was tested using a further 383 UK clinical isolates to detect signatures of recent foreign travel. Patient records indicated that in 46 cases, travel abroad had occurred <2 weeks prior to sampling, and genomic analysis identified that 34 (74%) of these isolates were of a non-UK origin. Identification of biogeographical markers in Campylobacter genomes will contribute to improved source attribution of clinical Campylobacter infection and inform intervention strategies to reduce campylobacteriosis.
Keywords: Campylobacter; allopatry; genomics; phylogeny; recombination; source attribution.
© 2017 The Authors. Molecular Ecology Published by John Wiley & Sons Ltd.
Figures
Similar articles
-
Genome-Wide Identification of Host-Segregating Epidemiological Markers for Source Attribution in Campylobacter jejuni.Appl Environ Microbiol. 2017 Mar 17;83(7):e03085-16. doi: 10.1128/AEM.03085-16. Print 2017 Apr 1. Appl Environ Microbiol. 2017. PMID: 28115376 Free PMC article.
-
Host association of Campylobacter genotypes transcends geographic variation.Appl Environ Microbiol. 2010 Aug;76(15):5269-77. doi: 10.1128/AEM.00124-10. Epub 2010 Jun 4. Appl Environ Microbiol. 2010. PMID: 20525862 Free PMC article.
-
Real-time genomic epidemiological evaluation of human Campylobacter isolates by use of whole-genome multilocus sequence typing.J Clin Microbiol. 2013 Aug;51(8):2526-34. doi: 10.1128/JCM.00066-13. Epub 2013 May 22. J Clin Microbiol. 2013. PMID: 23698529 Free PMC article.
-
A systematic review of source attribution of human campylobacteriosis using multilocus sequence typing.Euro Surveill. 2019 Oct;24(43):1800696. doi: 10.2807/1560-7917.ES.2019.24.43.1800696. Euro Surveill. 2019. PMID: 31662159 Free PMC article.
-
Pathogenomics of Emerging Campylobacter Species.Clin Microbiol Rev. 2019 Jul 3;32(4):e00072-18. doi: 10.1128/CMR.00072-18. Print 2019 Sep 18. Clin Microbiol Rev. 2019. PMID: 31270126 Free PMC article. Review.
Cited by
-
Gene pool transmission of multidrug resistance among Campylobacter from livestock, sewage and human disease.Environ Microbiol. 2019 Dec;21(12):4597-4613. doi: 10.1111/1462-2920.14760. Epub 2019 Aug 27. Environ Microbiol. 2019. PMID: 31385413 Free PMC article.
-
Genomic Relatedness, Antibiotic Resistance and Virulence Traits of Campylobacter jejuni HS19 Isolates From Cattle in China Indicate Pathogenic Potential.Front Microbiol. 2021 Nov 30;12:783750. doi: 10.3389/fmicb.2021.783750. eCollection 2021. Front Microbiol. 2021. PMID: 34956150 Free PMC article.
-
Machine learning to attribute the source of Campylobacter infections in the United States: A retrospective analysis of national surveillance data.J Infect. 2024 Nov;89(5):106265. doi: 10.1016/j.jinf.2024.106265. Epub 2024 Sep 7. J Infect. 2024. PMID: 39245152 Free PMC article.
-
Genome-wide identification of geographical segregated genetic markers in Salmonella enterica serovar Typhimurium variant 4,[5],12:i:Sci Rep. 2018 Oct 15;8(1):15251. doi: 10.1038/s41598-018-33266-5. Sci Rep. 2018. PMID: 30323193 Free PMC article.
-
Population Biology and Comparative Genomics of Campylobacter Species.Curr Top Microbiol Immunol. 2021;431:59-78. doi: 10.1007/978-3-030-65481-8_3. Curr Top Microbiol Immunol. 2021. PMID: 33620648 Review.
References
-
- Achtman, M. (2008). Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annual Review of Microbiology, 62, 53–70. - PubMed
-
- Cody, A. J. , Clarke, L. , Bowler, I. C. , & Dingle, K. E. (2010). Ciprofloxacin‐resistant campylobacteriosis in the UK. Lancet, 376, 1987. - PubMed
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
