

New insights into the role of mitochondrial calcium homeostasis in cell migration
- PMID: 28495532
- PMCID: PMC5930976
- DOI: 10.1016/j.bbrc.2017.05.039
New insights into the role of mitochondrial calcium homeostasis in cell migration
Abstract
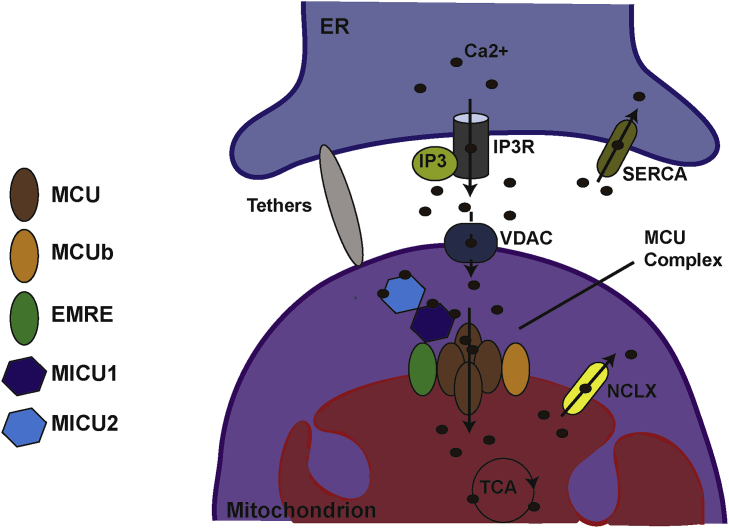
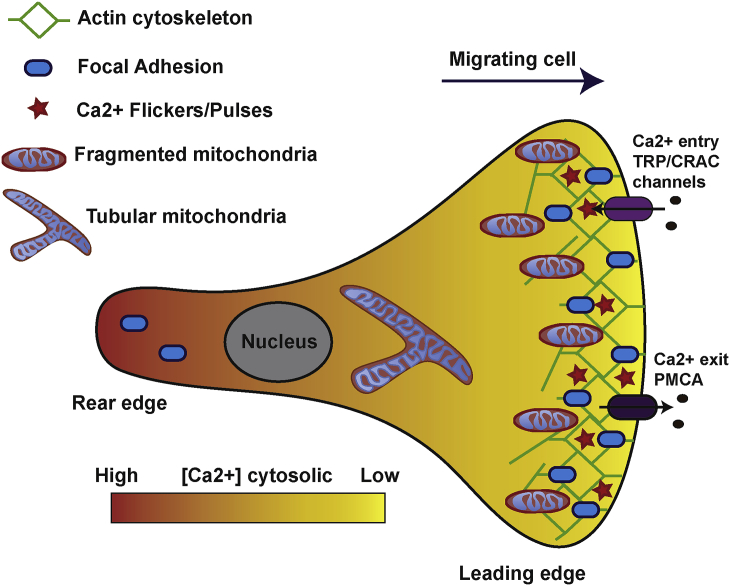
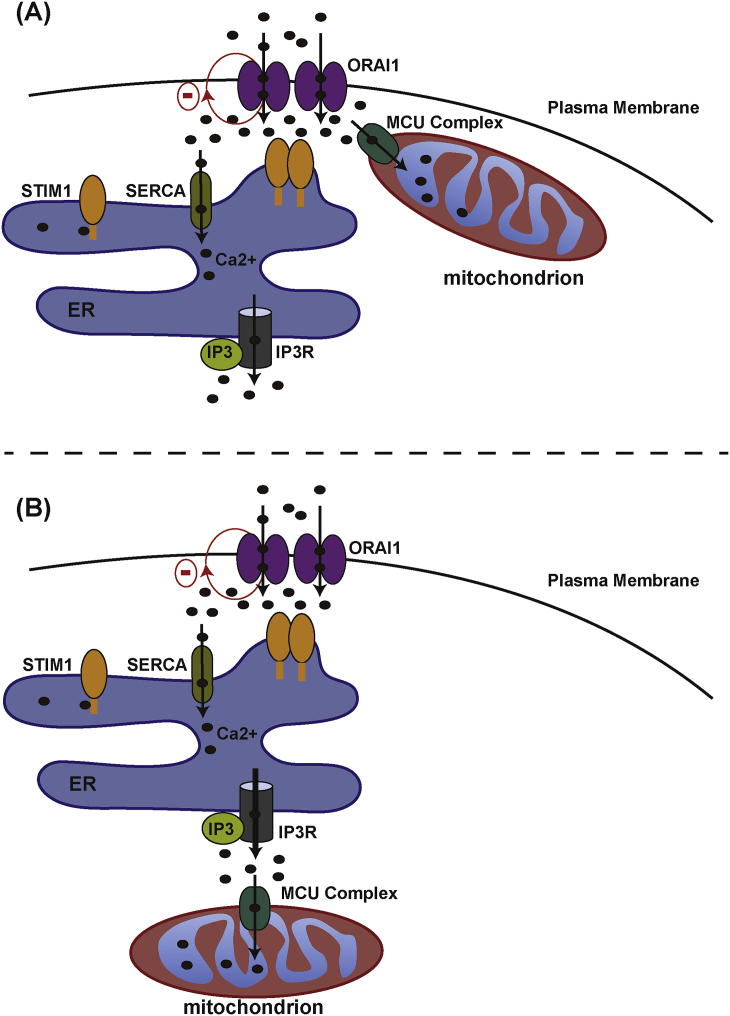
Mitochondria are dynamic organelles involved in numerous physiological functions. Beyond their function in ATP production, mitochondria regulate cell death, reactive oxygen species (ROS) generation, immunity and metabolism. Mitochondria also play a key role in the buffering of cytosolic calcium, and calcium transported into the matrix regulates mitochondrial metabolism. Recently, the identification of the mitochondrial calcium uniporter (MCU) and associated regulators has allowed the characterization of new physiological roles for calcium in both mitochondrial and cellular homeostasis. Indeed, recent work has highlighted the importance of mitochondrial calcium homeostasis in regulating cell migration. Cell migration is a property common to all metazoans and is critical to embryogenesis, cancer progression, wound-healing and immune surveillance. Previous work has established that cytoplasmic calcium is a key regulator of cell migration, as oscillations in cytosolic calcium activate cytoskeletal remodelling, actin contraction and focal adhesion (FA) turnover necessary for cell movement. Recent work using animal models and in cellulo experiments to genetically modulate MCU and partners have shed new light on the role of mitochondrial calcium dynamics in cytoskeletal remodelling through the modulation of ATP and ROS production, as well as intracellular calcium signalling. This review focuses on MCU and its regulators in cell migration during physiological and pathophysiological processes including development and cancer. We also present hypotheses to explain the molecular mechanisms by which MCU may regulate mitochondrial dynamics and motility to drive cell migration.
Keywords: Calcium; Cell migration; MCU; Mitochondria.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Molecular nature and physiological role of the mitochondrial calcium uniporter channel.Am J Physiol Cell Physiol. 2021 Apr 1;320(4):C465-C482. doi: 10.1152/ajpcell.00502.2020. Epub 2020 Dec 9. Am J Physiol Cell Physiol. 2021. PMID: 33296287 Free PMC article. Review.
-
MCU overexpression evokes disparate dose-dependent effects on mito-ROS and spontaneous Ca2+ release in hypertrophic rat cardiomyocytes.Am J Physiol Heart Circ Physiol. 2021 Oct 1;321(4):H615-H632. doi: 10.1152/ajpheart.00126.2021. Epub 2021 Aug 20. Am J Physiol Heart Circ Physiol. 2021. PMID: 34415186 Free PMC article.
-
CaMKII (Ca2+/Calmodulin-Dependent Kinase II) in Mitochondria of Smooth Muscle Cells Controls Mitochondrial Mobility, Migration, and Neointima Formation.Arterioscler Thromb Vasc Biol. 2018 Jun;38(6):1333-1345. doi: 10.1161/ATVBAHA.118.310951. Epub 2018 Mar 29. Arterioscler Thromb Vasc Biol. 2018. PMID: 29599132 Free PMC article.
-
MCU-i4, a mitochondrial Ca2+ uniporter modulator, induces breast cancer BT474 cell death by enhancing glycolysis, ATP production and reactive oxygen species (ROS) burst.Oncol Res. 2025 Jan 16;33(2):397-406. doi: 10.32604/or.2024.052743. eCollection 2025. Oncol Res. 2025. PMID: 39866241 Free PMC article.
-
Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes.Trends Biochem Sci. 2016 Dec;41(12):1035-1049. doi: 10.1016/j.tibs.2016.09.001. Epub 2016 Sep 28. Trends Biochem Sci. 2016. PMID: 27692849 Free PMC article. Review.
Cited by
-
Optimizing Calcium Detection Methods in Animal Systems: A Sandbox for Synthetic Biology.Biomolecules. 2021 Feb 24;11(3):343. doi: 10.3390/biom11030343. Biomolecules. 2021. PMID: 33668387 Free PMC article. Review.
-
Roles of mitochondria in neutrophils.Front Immunol. 2022 Aug 19;13:934444. doi: 10.3389/fimmu.2022.934444. eCollection 2022. Front Immunol. 2022. PMID: 36081497 Free PMC article. Review.
-
Genetic Ablation of the Mitochondrial Calcium Uniporter (MCU) Does not Impair T Cell-Mediated Immunity In Vivo.Front Pharmacol. 2021 Dec 20;12:734078. doi: 10.3389/fphar.2021.734078. eCollection 2021. Front Pharmacol. 2021. PMID: 34987384 Free PMC article.
-
Biological Aging and the Cellular Pathogenesis of Huntington's Disease.J Huntingtons Dis. 2020;9(2):115-128. doi: 10.3233/JHD-200395. J Huntingtons Dis. 2020. PMID: 32417788 Free PMC article. Review.
-
A Putative Prohibitin-Calcium Nexus in β-Cell Mitochondria and Diabetes.J Diabetes Res. 2020 Oct 8;2020:7814628. doi: 10.1155/2020/7814628. eCollection 2020. J Diabetes Res. 2020. PMID: 33354575 Free PMC article. Review.
References
-
- Naon D., Scorrano L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Biophys. Acta. 2014 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
