

Mutations in COL1A1 and COL1A2 and dental aberrations in children and adolescents with osteogenesis imperfecta - A retrospective cohort study
- PMID: 28498836
- PMCID: PMC5428910
- DOI: 10.1371/journal.pone.0176466
Mutations in COL1A1 and COL1A2 and dental aberrations in children and adolescents with osteogenesis imperfecta - A retrospective cohort study
Abstract
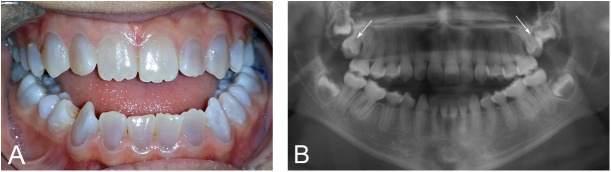
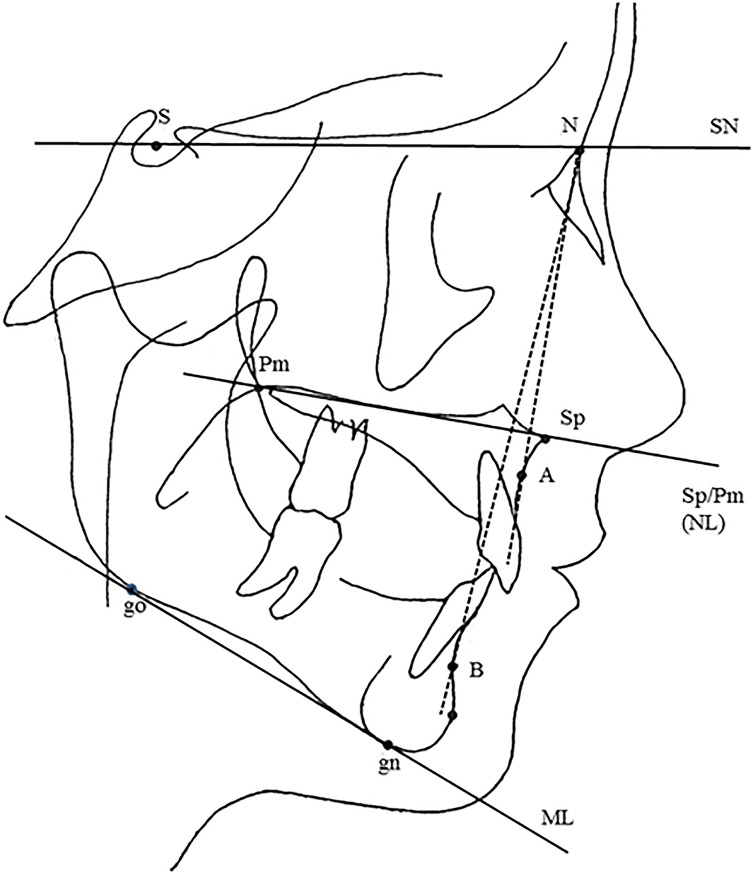
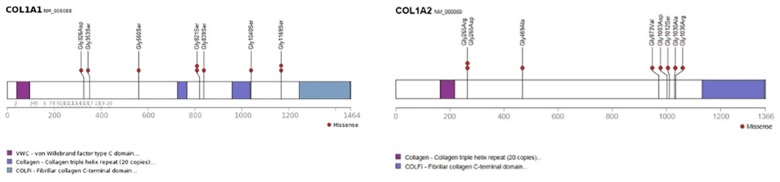
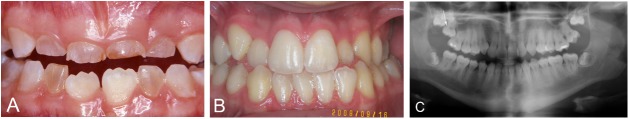
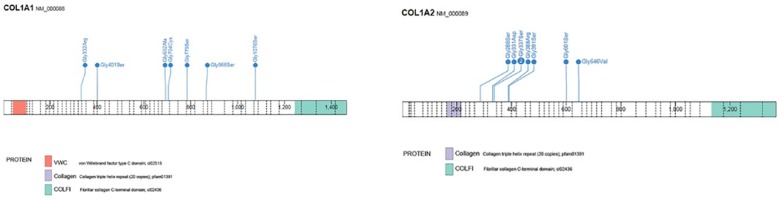
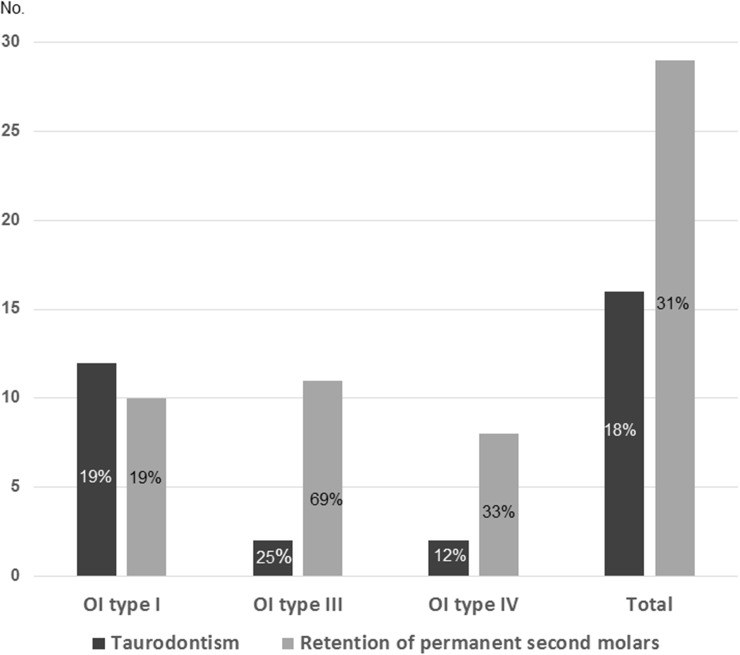
Osteogenesis imperfecta (OI) is a heterogeneous group of disorders of connective tissue, caused mainly by mutations in the collagen I genes (COL1A1 and COL1A2). Dentinogenesis imperfecta (DGI) and other dental aberrations are common features of OI. We investigated the association between collagen I mutations and DGI, taurodontism, and retention of permanent second molars in a retrospective cohort of 152 unrelated children and adolescents with OI. The clinical examination included radiographic evaluations. Teeth from 81 individuals were available for histopathological evaluation. COL1A1/2 mutations were found in 104 individuals by nucleotide sequencing. DGI was diagnosed clinically and radiographically in 29% of the individuals (44/152) and through isolated histological findings in another 19% (29/152). In the individuals with a COL1A1 mutation, 70% (7/10) of those with a glycine substitution located C-terminal of p.Gly305 exhibited DGI in both dentitions while no individual (0/7) with a mutation N-terminal of this point exhibited DGI in either dentition (p = 0.01). In the individuals with a COL1A2 mutation, 80% (8/10) of those with a glycine substitution located C terminal of p.Gly211 exhibited DGI in both dentitions while no individual (0/5) with a mutation N-terminal of this point (p = 0.007) exhibited DGI in either dentition. DGI was restricted to the deciduous dentition in 20 individuals. Seventeen had missense mutations where glycine to serine was the most prevalent substitution (53%). Taurodontism occurred in 18% and retention of permanent second molars in 31% of the adolescents. Dental aberrations are strongly associated with qualitatively changed collagen I. The varying expressivity of DGI is related to the location of the collagen I mutation. Genotype information may be helpful in identifying individuals with OI who have an increased risk of dental aberrations.
Conflict of interest statement
Figures

References
-
- Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28(3):209–21. PubMed Central PMCID: PMCPMC4144349. doi: 10.1002/humu.20429 - DOI - PMC - PubMed
-
- Sykes B, Ogilvie D, Wordsworth P, Anderson, Jones N. Osteogenesis imperfecta is linked to both type I collagen structural genes. Lancet. 1986;2(8498):69–72. - PubMed
-
- Bardai G, Moffatt P, Glorieux FH, Rauch F. DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: diagnostic yield and mutation spectrum. Osteoporos Int. 2016. - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
