

Resistance to RET-Inhibition in RET-Rearranged NSCLC Is Mediated By Reactivation of RAS/MAPK Signaling
- PMID: 28500237
- PMCID: PMC5544556
- DOI: 10.1158/1535-7163.MCT-17-0008
Resistance to RET-Inhibition in RET-Rearranged NSCLC Is Mediated By Reactivation of RAS/MAPK Signaling
Abstract
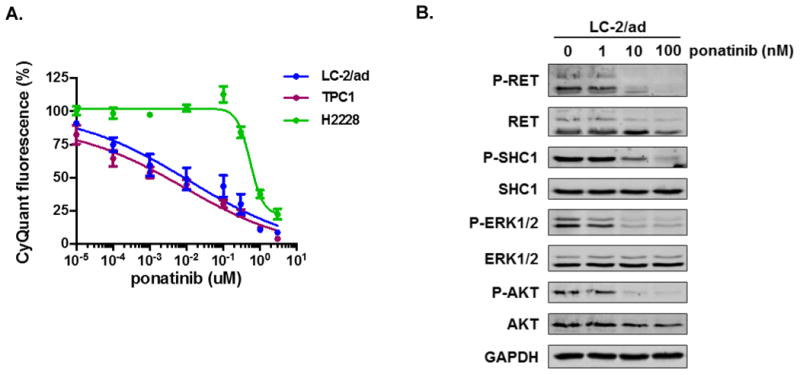
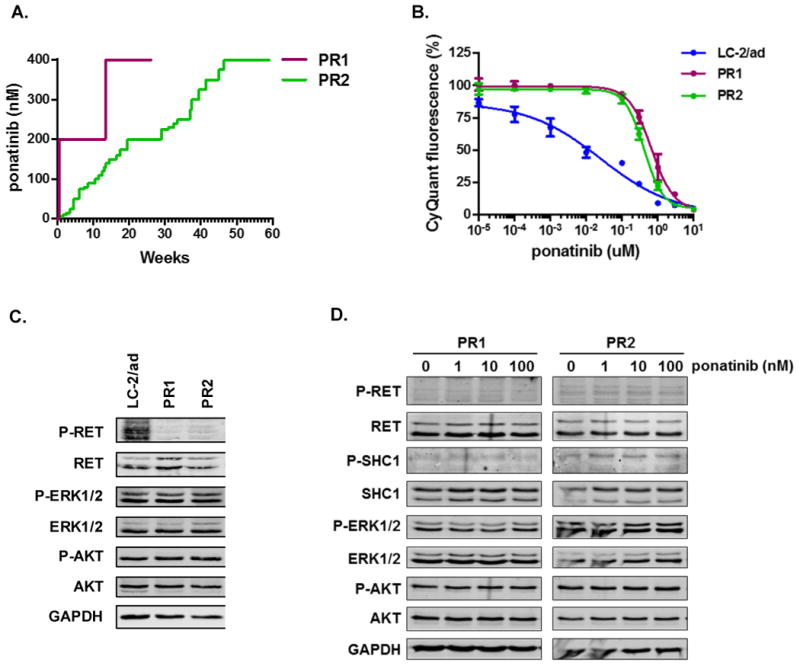
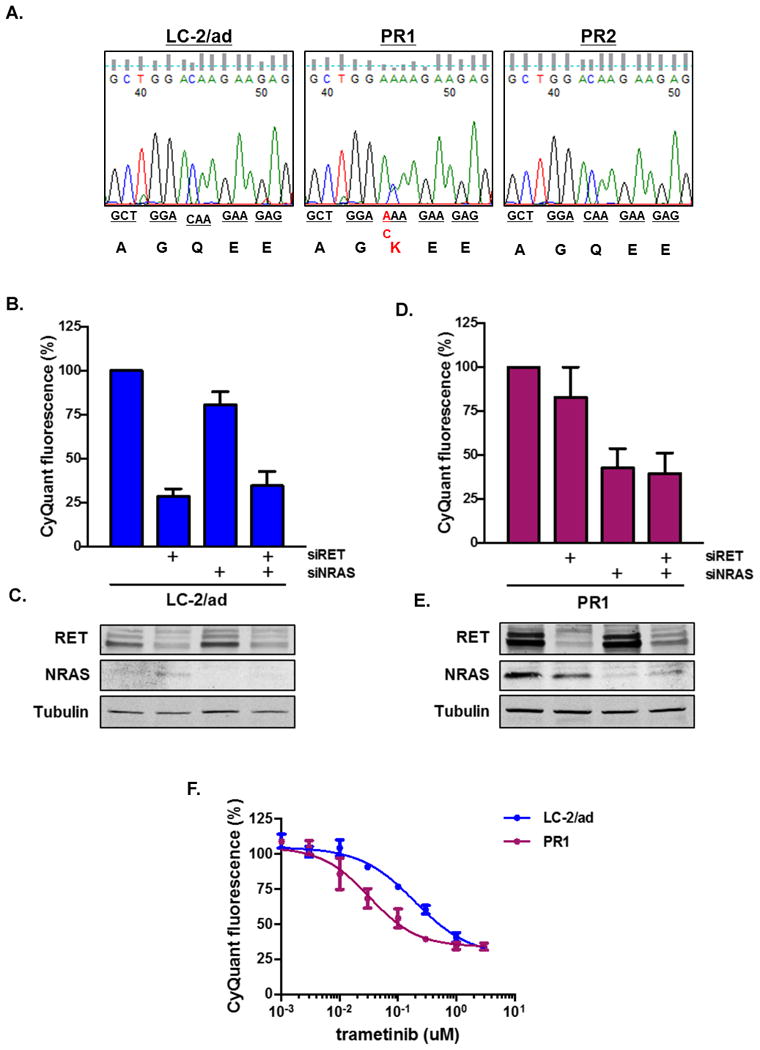
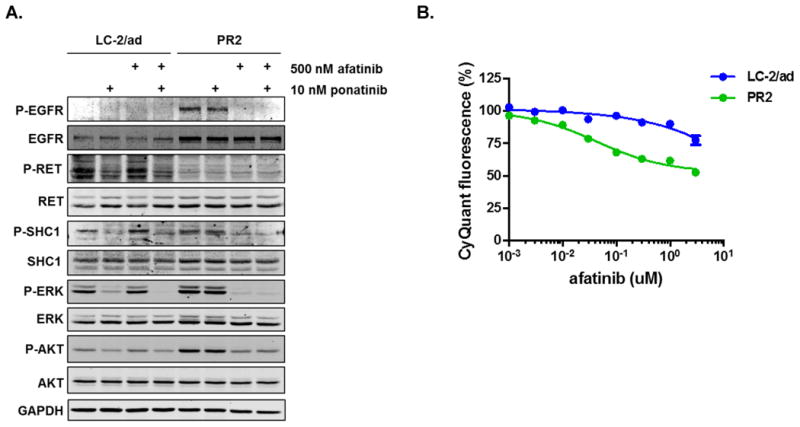
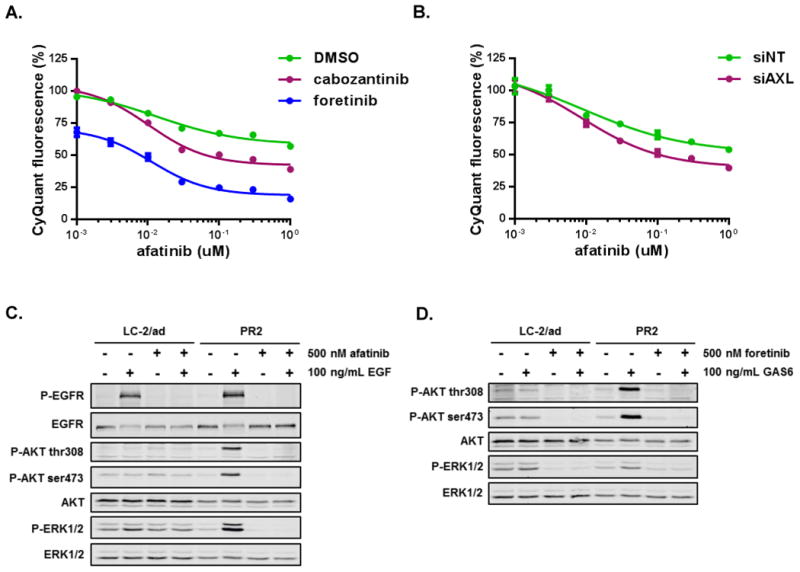
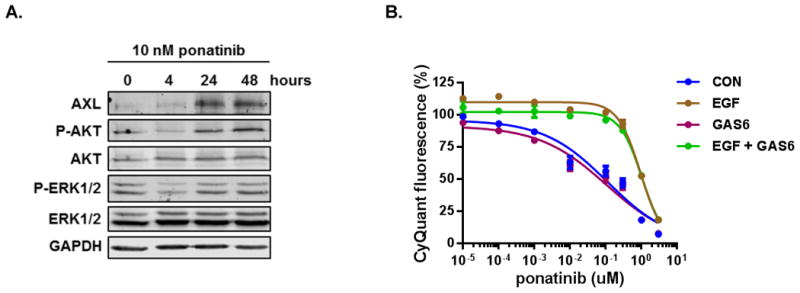
Oncogenic rearrangements in RET are present in 1%-2% of lung adenocarcinoma patients. Ponatinib is a multi-kinase inhibitor with low-nanomolar potency against the RET kinase domain. Here, we demonstrate that ponatinib exhibits potent antiproliferative activity in RET fusion-positive LC-2/ad lung adenocarcinoma cells and inhibits phosphorylation of the RET fusion protein and signaling through ERK1/2 and AKT. Using distinct dose escalation strategies, two ponatinib-resistant LC-2/ad cell lines, PR1 and PR2, were derived. PR1 and PR2 cell lines retained expression, but not phosphorylation of the RET fusion and lacked evidence of a resistance mutation in the RET kinase domain. Both resistant lines retained activation of the MAPK pathway. Next-generation RNA sequencing revealed an oncogenic NRAS p.Q61K mutation in the PR1 cell. PR1 cell proliferation was preferentially sensitive to siRNA knockdown of NRAS compared with knockdown of RET, more sensitive to MEK inhibition than the parental line, and NRAS dependence was maintained in the absence of chronic RET inhibition. Expression of NRAS p.Q61K in RET fusion expressing TPC1 cells conferred resistance to ponatinib. PR2 cells exhibited increased expression of EGFR and AXL. EGFR inhibition decreased cell proliferation and phosphorylation of ERK1/2 and AKT in PR2 cells, but not LC-2/ad cells. Although AXL inhibition enhanced PR2 sensitivity to afatinib, it was unable to decrease cell proliferation by itself. Thus, EGFR and AXL cooperatively rescued signaling from RET inhibition in the PR2 cells. Collectively, these findings demonstrate that resistance to ponatinib in RET-rearranged lung adenocarcinoma is mediated by bypass signaling mechanisms that result in restored RAS/MAPK activation. Mol Cancer Ther; 16(8); 1623-33. ©2017 AACR.
©2017 American Association for Cancer Research.
Figures
Similar articles
-
MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation.Cancer Res. 2014 Jan 1;74(1):253-62. doi: 10.1158/0008-5472.CAN-13-1103. Epub 2013 Oct 28. Cancer Res. 2014. PMID: 24165158
-
EGF Induced RET Inhibitor Resistance in CCDC6-RET Lung Cancer Cells.Yonsei Med J. 2017 Jan;58(1):9-18. doi: 10.3349/ymj.2017.58.1.9. Yonsei Med J. 2017. PMID: 27873490 Free PMC article.
-
Ponatinib (AP24534) is a novel potent inhibitor of oncogenic RET mutants associated with thyroid cancer.J Clin Endocrinol Metab. 2013 May;98(5):E811-9. doi: 10.1210/jc.2012-2672. Epub 2013 Mar 22. J Clin Endocrinol Metab. 2013. PMID: 23526464
-
Treatment of Brain Metastases of Non-Small Cell Lung Carcinoma.Int J Mol Sci. 2021 Jan 8;22(2):593. doi: 10.3390/ijms22020593. Int J Mol Sci. 2021. PMID: 33435596 Free PMC article. Review.
-
Strategies to overcome acquired resistance to EGFR TKI in the treatment of non-small cell lung cancer.Clin Transl Oncol. 2019 Oct;21(10):1287-1301. doi: 10.1007/s12094-019-02075-1. Epub 2019 Mar 12. Clin Transl Oncol. 2019. PMID: 30864018 Review.
Cited by
-
Role of metabolic reprogramming in drug resistance to epidermal growth factor tyrosine kinase inhibitors in non-small cell lung cancer.Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2021 May 28;46(5):545-551. doi: 10.11817/j.issn.1672-7347.2021.200529. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2021. PMID: 34148892 Free PMC article. Chinese, English.
-
Activation of the AKT-mTOR pathway confers selpercatinib resistance in thyroid cancer cells harboring the CCDC6-RET fusion gene.Biochem Biophys Rep. 2025 Jul 7;43:102136. doi: 10.1016/j.bbrep.2025.102136. eCollection 2025 Sep. Biochem Biophys Rep. 2025. PMID: 40688508 Free PMC article.
-
Molecular genetics, therapeutics and RET inhibitor resistance for medullary thyroid carcinoma and future perspectives.Cell Commun Signal. 2024 Sep 28;22(1):460. doi: 10.1186/s12964-024-01837-x. Cell Commun Signal. 2024. PMID: 39342195 Free PMC article. Review.
-
SOS2 regulates the threshold of mutant EGFR-dependent oncogenesis.bioRxiv [Preprint]. 2023 Jun 29:2023.01.20.524989. doi: 10.1101/2023.01.20.524989. bioRxiv. 2023. Update in: Mol Oncol. 2024 Mar;18(3):641-661. doi: 10.1002/1878-0261.13564. PMID: 37425733 Free PMC article. Updated. Preprint.
-
A Rapid, Functional sgRNA Screening Method for Generating Murine RET and NTRK1 Fusion Oncogenes.bioRxiv [Preprint]. 2023 Apr 6:2023.04.06.535912. doi: 10.1101/2023.04.06.535912. bioRxiv. 2023. Update in: Biol Open. 2023 Aug 15;12(8):bio059994. doi: 10.1242/bio.059994. PMID: 37066347 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
