

DNA methylation and DNA methyltransferases
- PMID: 28503201
- PMCID: PMC5422929
- DOI: 10.1186/s13072-017-0130-8
DNA methylation and DNA methyltransferases
Abstract
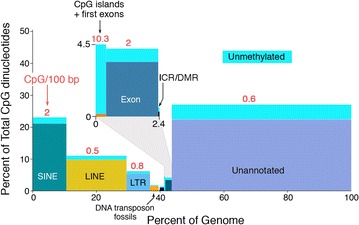
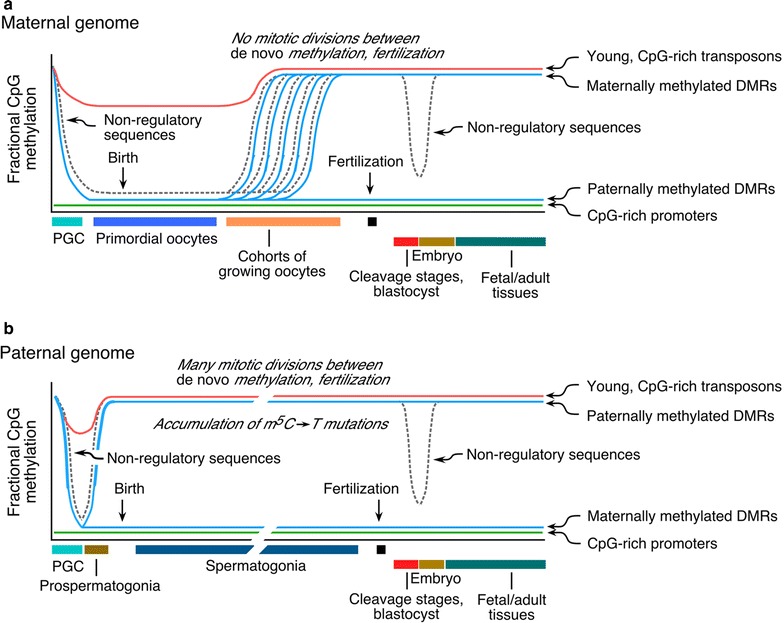
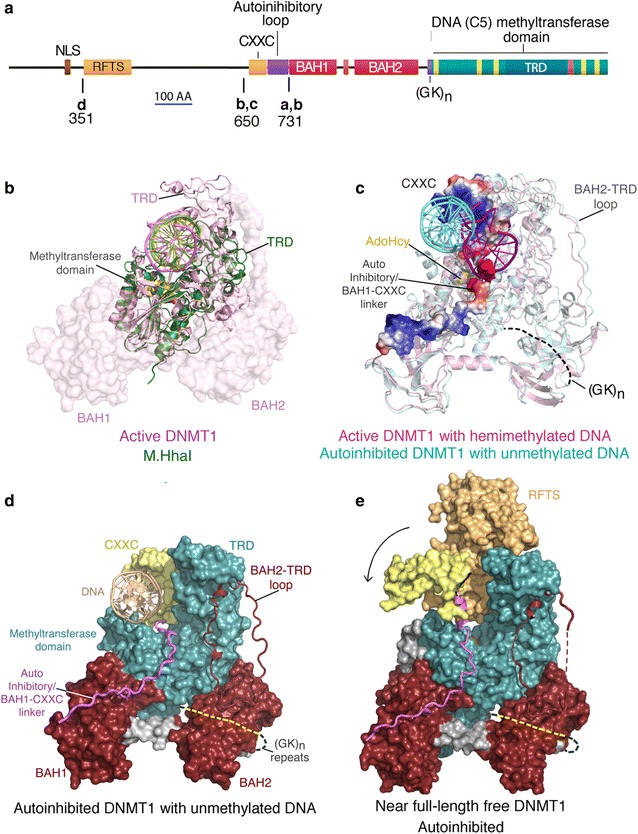
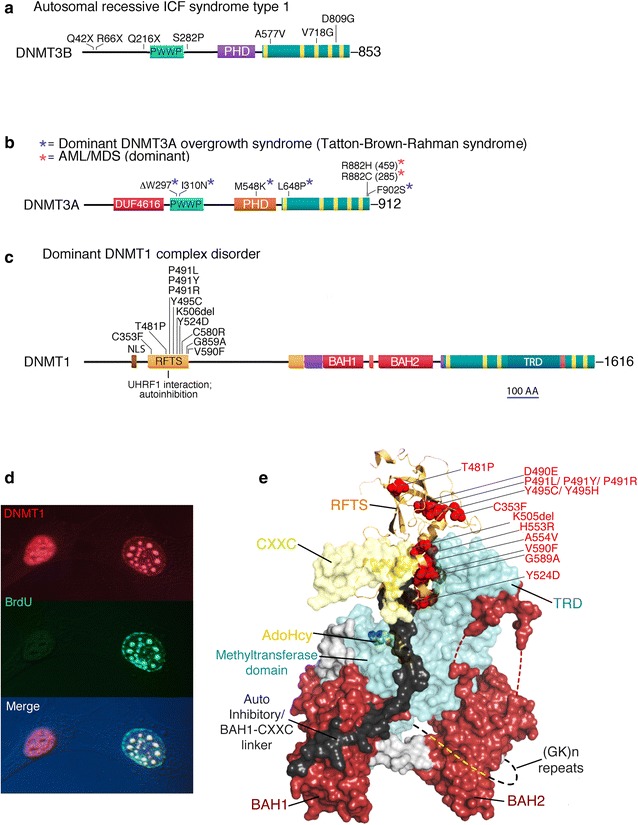
The prevailing views as to the form, function, and regulation of genomic methylation patterns have their origin many years in the past, at a time when the structure of the mammalian genome was only dimly perceived, when the number of protein-encoding mammalian genes was believed to be at least five times greater than the actual number, and when it was not understood that only ~10% of the genome is under selective pressure and likely to have biological function. We use more recent findings from genome biology and whole-genome methylation profiling to provide a reappraisal of the shape of genomic methylation patterns and the nature of the changes that they undergo during gametogenesis and early development. We observe that the sequences that undergo deep changes in methylation status during early development are largely sequences without regulatory function. We also discuss recent findings that begin to explain the remarkable fidelity of maintenance methylation. Rather than a general overview of DNA methylation in mammals (which has been the subject of many reviews), we present a new analysis of the distribution of methylated CpG dinucleotides across the multiple sequence compartments that make up the mammalian genome, and we offer an updated interpretation of the nature of the changes in methylation patterns that occur in germ cells and early embryos. We discuss the cues that might designate specific sequences for demethylation or de novo methylation during development, and we summarize recent findings on mechanisms that maintain methylation patterns in mammalian genomes. We also describe the several human disorders, each very different from the other, that are caused by mutations in DNA methyltransferase genes.
Keywords: DNA cytosine methylation; Epigenetics; Mammalian DNA methyltransferases; Methylation dynamics; Methylation-related human diseases.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
