

Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma
- PMID: 28504693
- PMCID: PMC5523066
- DOI: 10.1038/oncsis.2017.33
Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma
Abstract
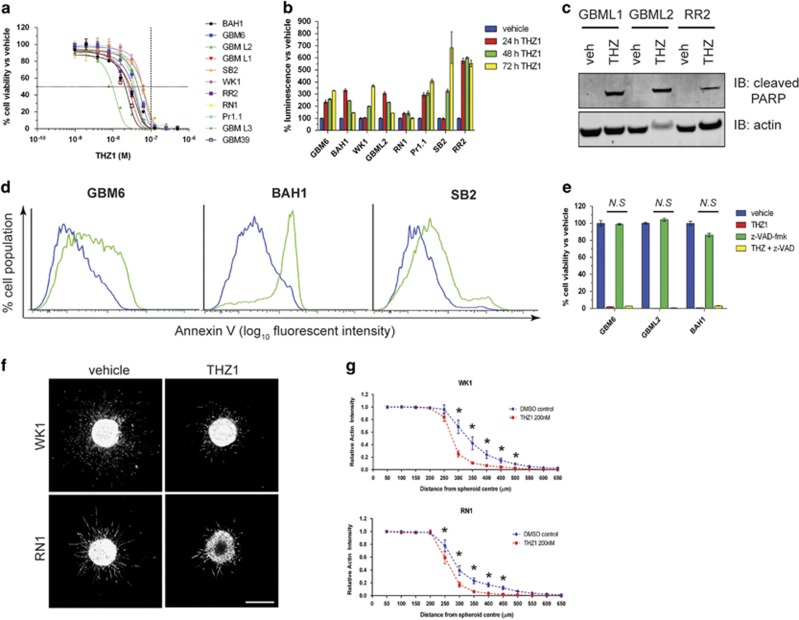
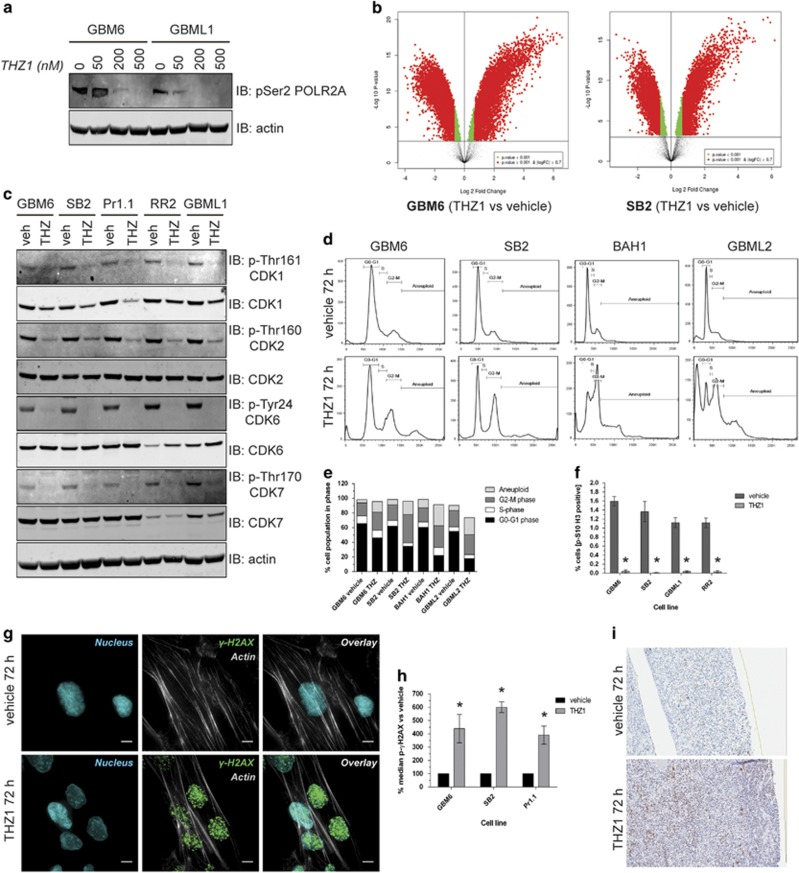
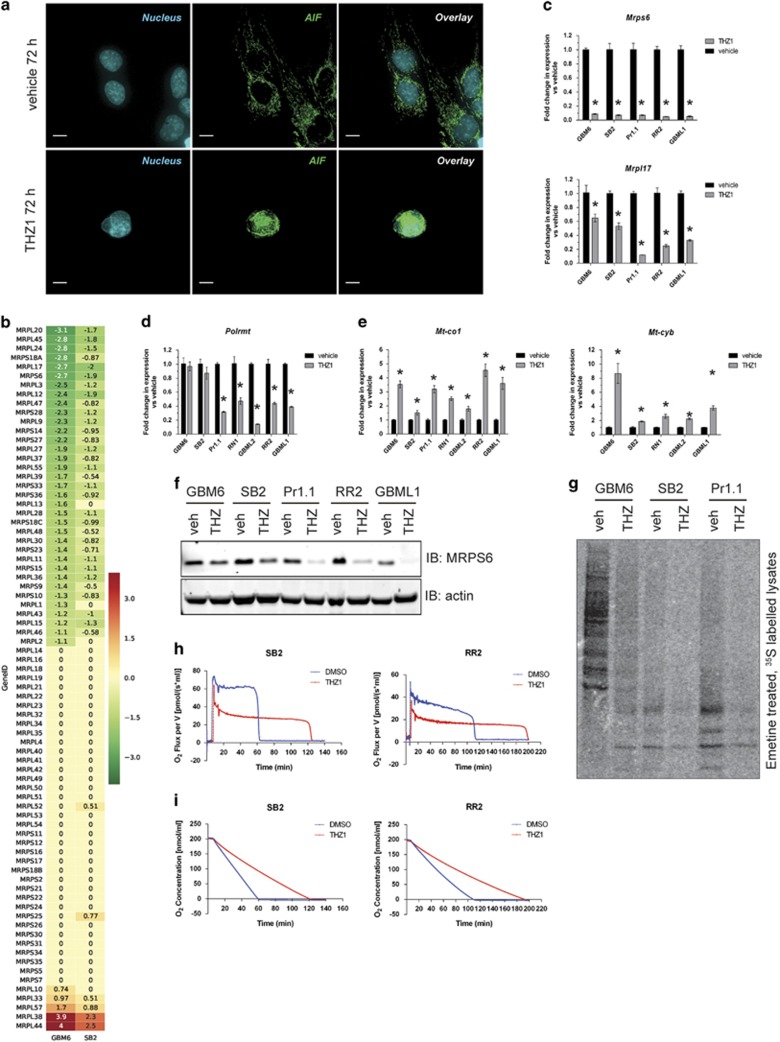
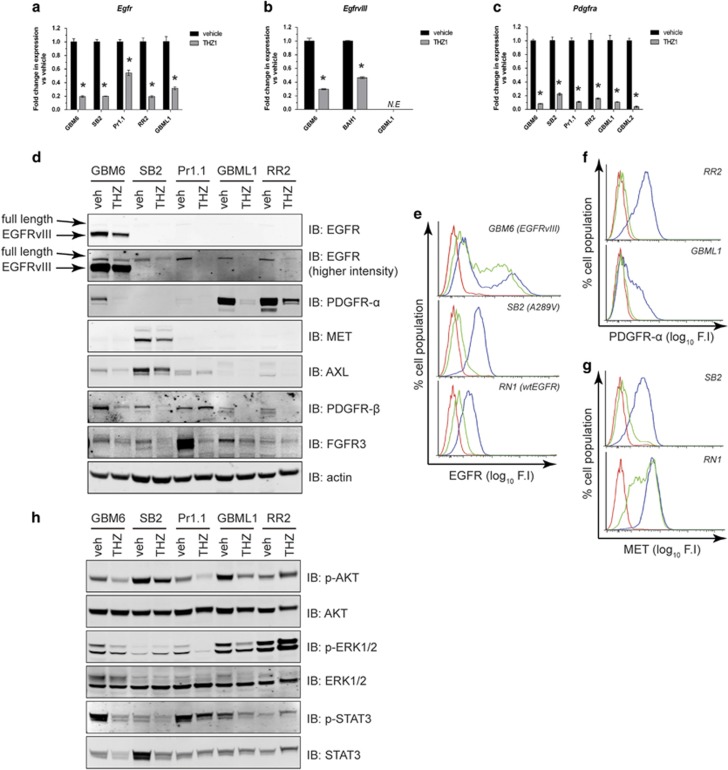
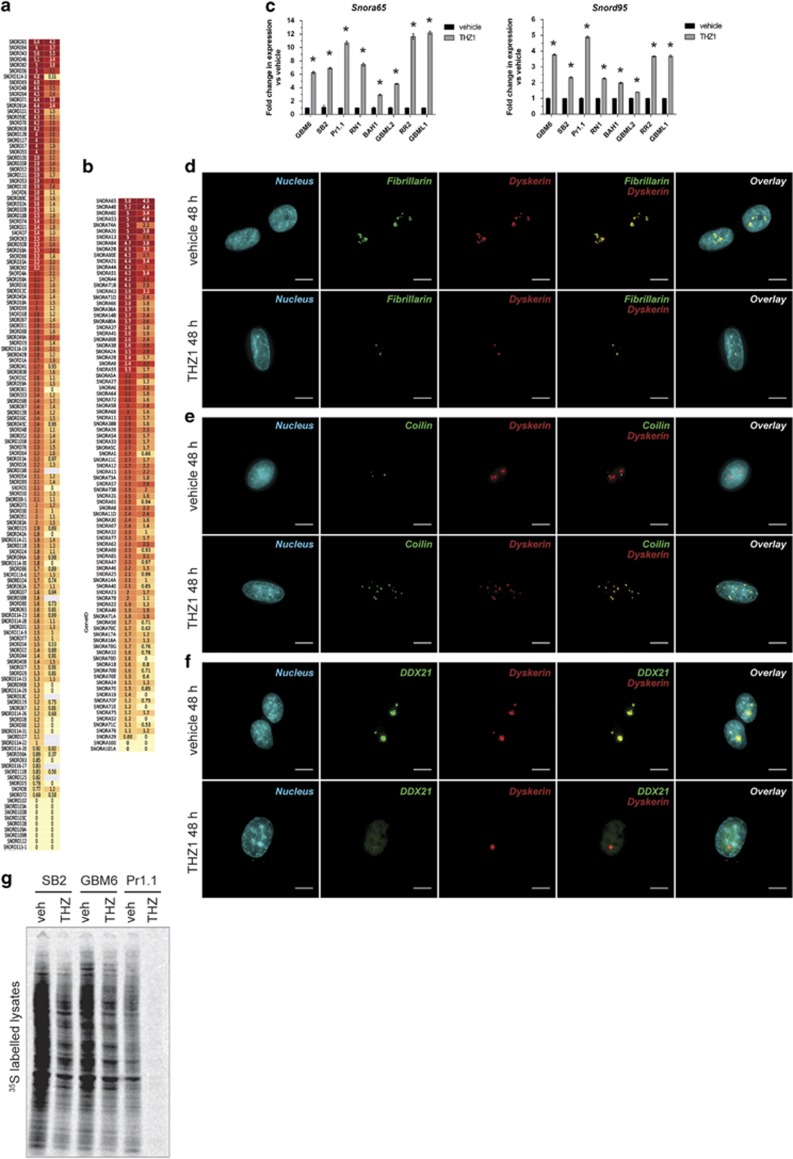
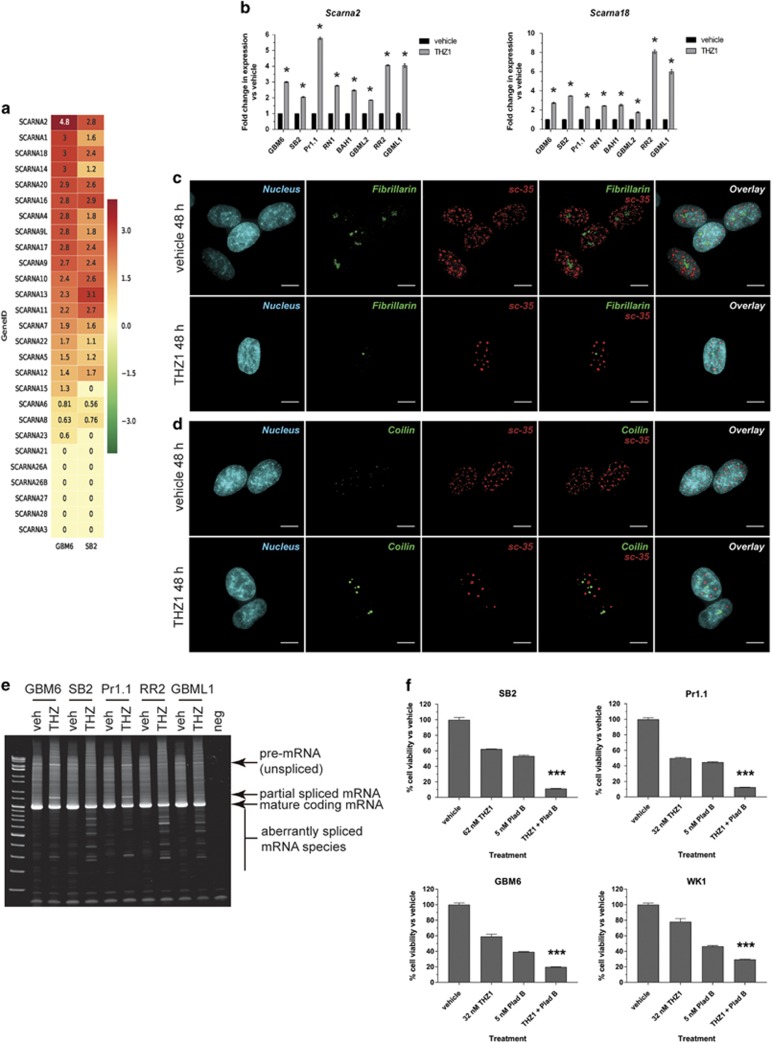
High-grade glioma (HGG) is an incurable brain cancer. The transcriptomes of cells within HGG tumors are highly heterogeneous. This renders the tumors unresponsive or able to adapt to therapeutics targeted at single pathways, thereby causing treatment failure. To overcome this, we focused on cyclin-dependent kinase 7 (CDK7), a ubiquitously expressed molecule involved in two major drivers of HGG pathogenesis: cell cycle progression and RNA polymerase-II-based transcription. We tested the activity of THZ1, an irreversible CDK7 inhibitor, on patient-derived primary HGG cell lines and ex vivo HGG patient tissue slices, using proliferation assays, microarray analysis, high-resolution respirometry, cell cycle analysis and in vivo tumor orthografts. The cellular processes affected by CDK7 inhibition were analyzed by reverse transcriptase-quantitative PCR, western blot, flow cytometry and immunofluorescence. THZ1 perturbed the transcriptome and disabled CDK activation, leading to cell cycle arrest at G2 and DNA damage. THZ1 halted transcription of the nuclear-encoded mitochondrial ribosomal genes, reducing mitochondrial translation and oxidative respiration. It also inhibited the expression of receptor tyrosine kinases such as epidermal growth factor receptor (EGFR) and platelet-derived growth factor receptor-α (PDGFR-α), reducing signaling flux through the AKT, extracellular-signal-regulated kinase 1/2 (ERK1/2) and signal transducer and activator of transcription 3 (STAT3) downstream pathways. Finally, THZ1 disrupted nucleolar, Cajal body and nuclear speckle formation, resulting in reduced cytosolic translation and malfunction of the spliceosome and thus leading to aberrant mRNA processing. These findings indicate that CDK7 is crucial for gliomagenesis, validate CDK7 as a therapeutic target and provide new insight into the cellular processes that are affected by THZ1 and induce antitumor activity.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011; 20: 810–817. - PubMed
-
- Kim J, Lee IH, Cho HJ, Park CK, Jung YS, Kim Y et al. Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell 2015; 28: 318–328. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
