

Unveiling a novel transient druggable pocket in BACE-1 through molecular simulations: Conformational analysis and binding mode of multisite inhibitors
- PMID: 28505196
- PMCID: PMC5432175
- DOI: 10.1371/journal.pone.0177683
Unveiling a novel transient druggable pocket in BACE-1 through molecular simulations: Conformational analysis and binding mode of multisite inhibitors
Erratum in
-
Correction: Unveiling a novel transient druggable pocket in BACE-1 through molecular simulations: Conformational analysis and binding mode of multisite inhibitors.PLoS One. 2017 Dec 21;12(12):e0190327. doi: 10.1371/journal.pone.0190327. eCollection 2017. PLoS One. 2017. PMID: 29267385 Free PMC article.
Abstract
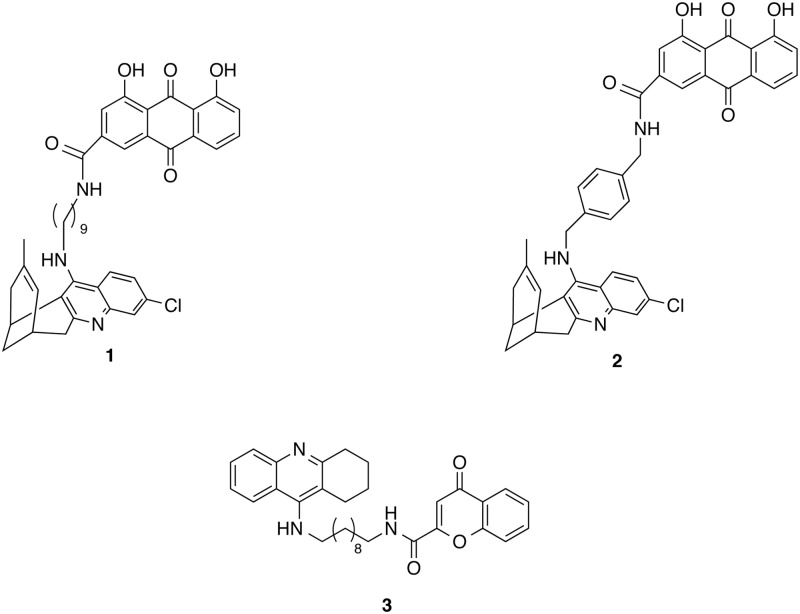
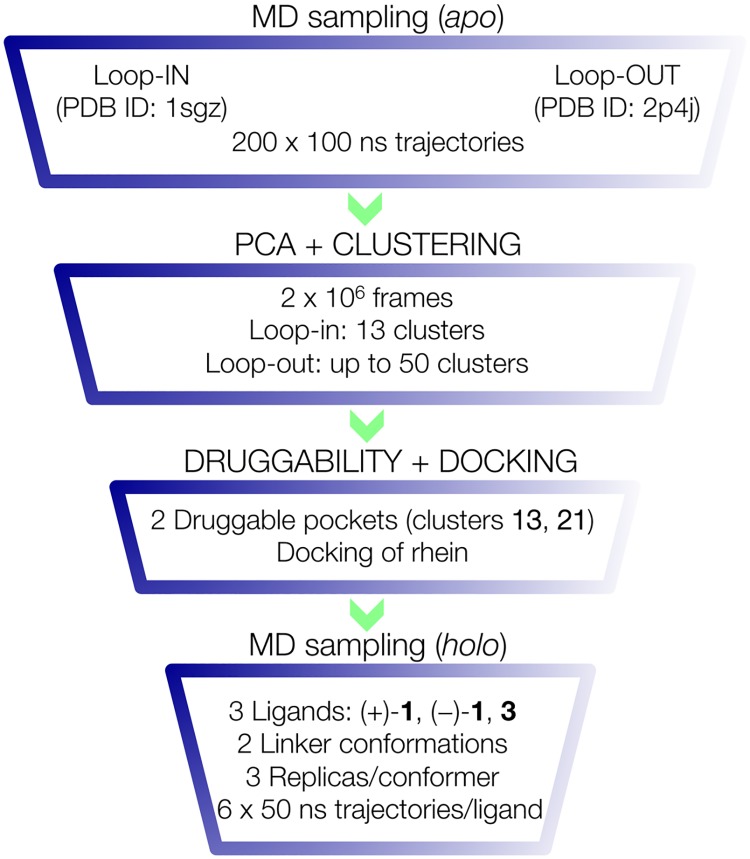
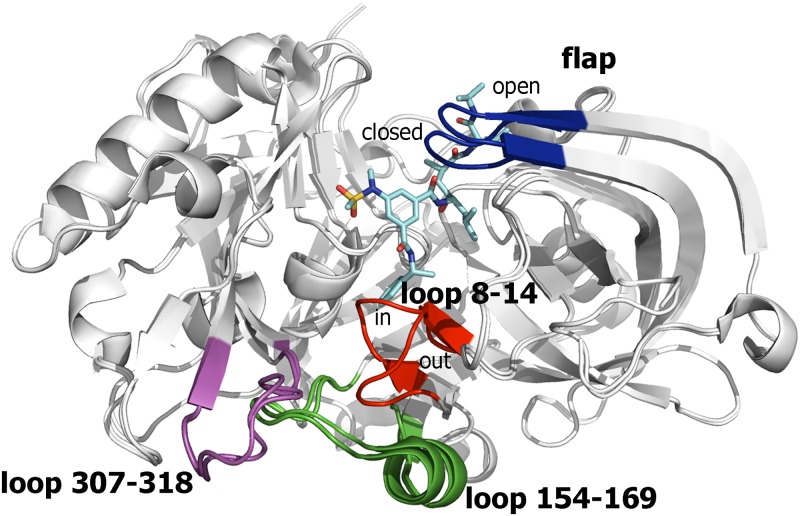
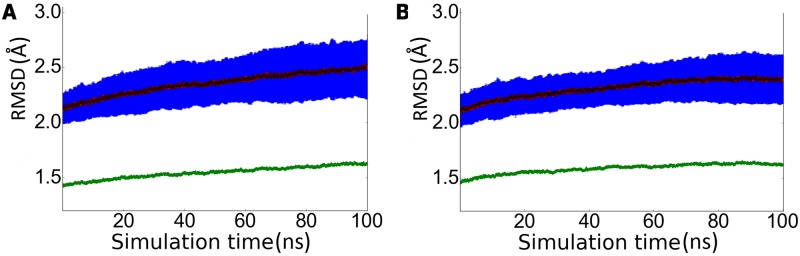
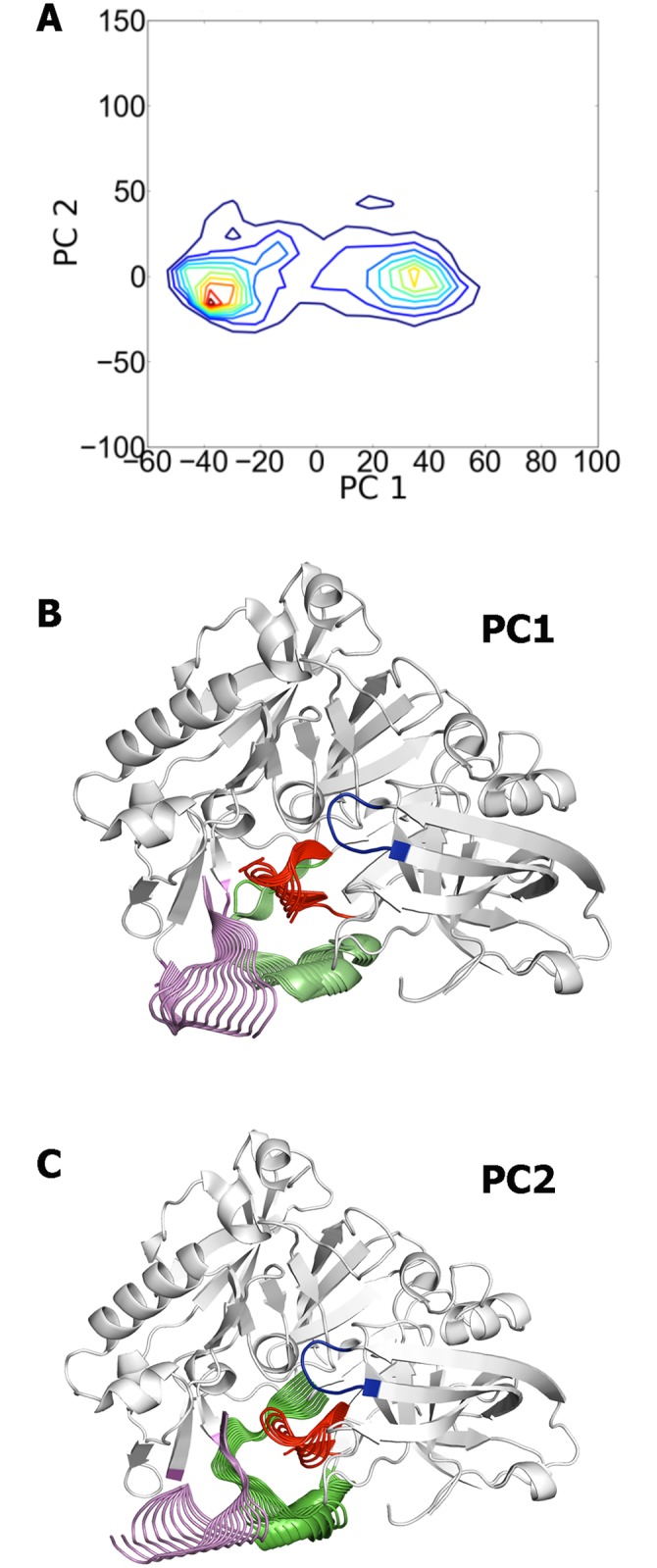
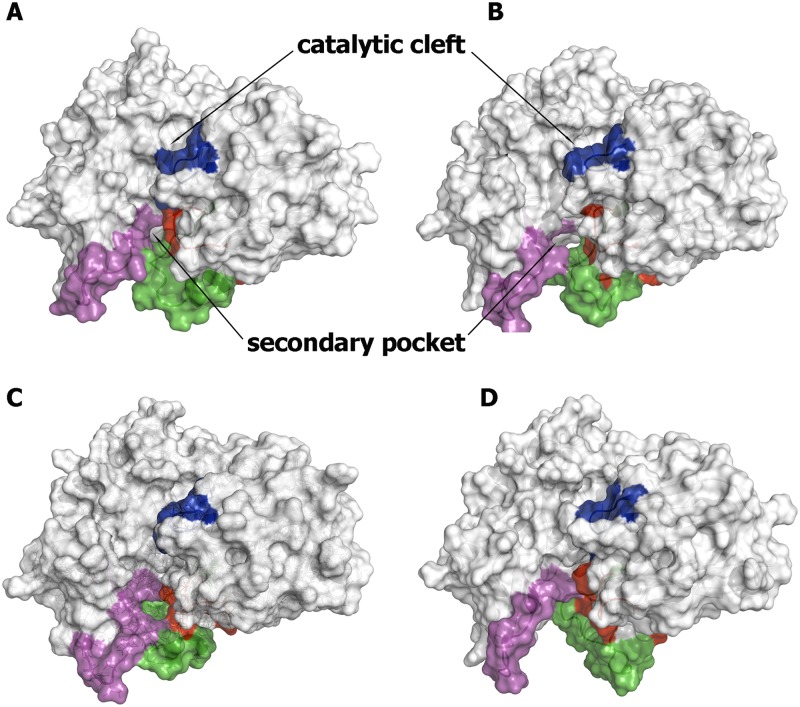
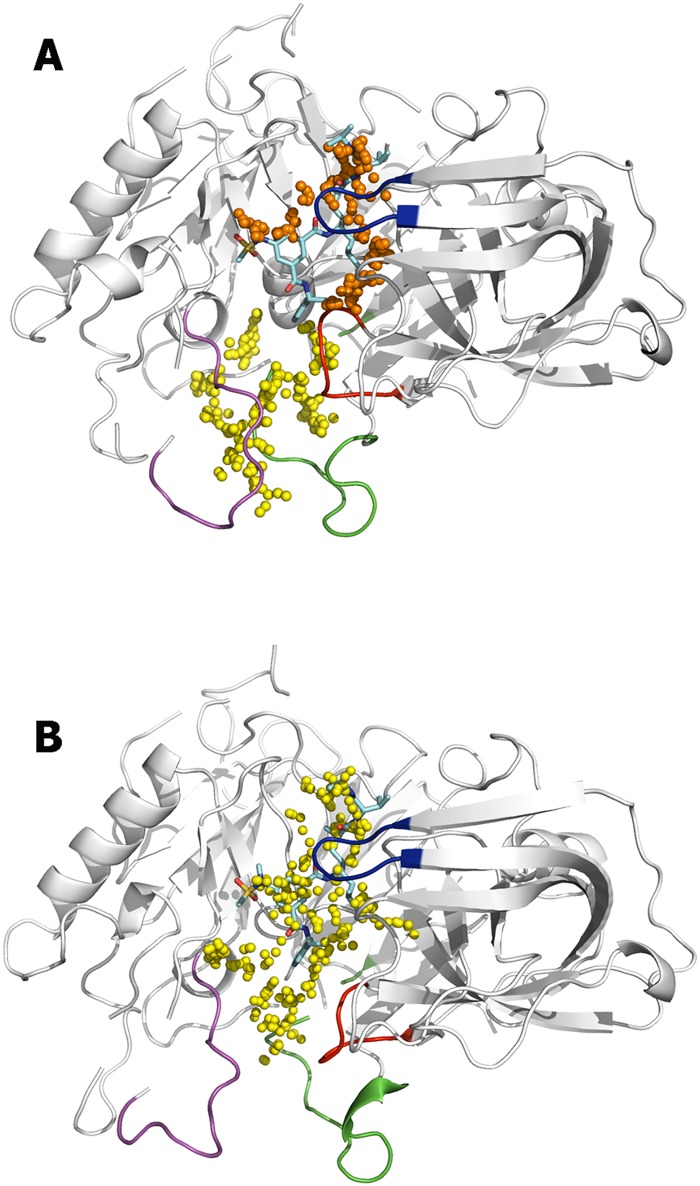
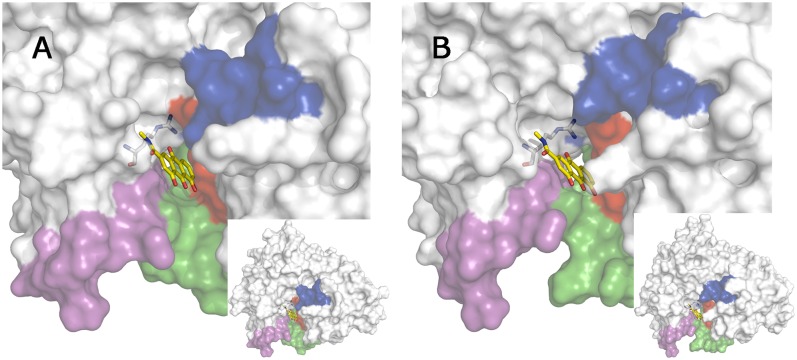
The critical role of BACE-1 in the formation of neurotoxic ß-amyloid peptides in the brain makes it an attractive target for an efficacious treatment of Alzheimer's disease. However, the development of clinically useful BACE-1 inhibitors has proven to be extremely challenging. In this study we examine the binding mode of a novel potent inhibitor (compound 1, with IC50 80 nM) designed by synergistic combination of two fragments-huprine and rhein-that individually are endowed with very low activity against BACE-1. Examination of crystal structures reveals no appropriate binding site large enough to accommodate 1. Therefore we have examined the conformational flexibility of BACE-1 through extended molecular dynamics simulations, paying attention to the highly flexible region shaped by loops 8-14, 154-169 and 307-318. The analysis of the protein dynamics, together with studies of pocket druggability, has allowed us to detect the transient formation of a secondary binding site, which contains Arg307 as a key residue for the interaction with small molecules, at the edge of the catalytic cleft. The formation of this druggable "floppy" pocket would enable the binding of multisite inhibitors targeting both catalytic and secondary sites. Molecular dynamics simulations of BACE-1 bound to huprine-rhein hybrid compounds support the feasibility of this hypothesis. The results provide a basis to explain the high inhibitory potency of the two enantiomeric forms of 1, together with the large dependence on the length of the oligomethylenic linker. Furthermore, the multisite hypothesis has allowed us to rationalize the inhibitory potency of a series of tacrine-chromene hybrid compounds, specifically regarding the apparent lack of sensitivity of the inhibition constant to the chemical modifications introduced in the chromene unit. Overall, these findings pave the way for the exploration of novel functionalities in the design of optimized BACE-1 multisite inhibitors.
Conflict of interest statement
Figures
Similar articles
-
Docking, molecular dynamics, binding energy-MM-PBSA studies of naphthofuran derivatives to identify potential dual inhibitors against BACE-1 and GSK-3β.J Biomol Struct Dyn. 2019 Feb;37(2):275-290. doi: 10.1080/07391102.2018.1426043. Epub 2018 Jan 19. J Biomol Struct Dyn. 2019. PMID: 29310523
-
Study on the active mechanism of β-secretase inhibitors by molecular simulations.Eur J Pharm Sci. 2015 Aug 30;76:138-48. doi: 10.1016/j.ejps.2015.05.007. Epub 2015 May 9. Eur J Pharm Sci. 2015. PMID: 25965961
-
Design and synthesis of novel 3,5-bis-N-(aryl/heteroaryl) carbamoyl-4-aryl-1,4-dihydropyridines as small molecule BACE-1 inhibitors.Bioorg Med Chem. 2013 Nov 15;21(22):6893-909. doi: 10.1016/j.bmc.2013.09.033. Epub 2013 Sep 20. Bioorg Med Chem. 2013. PMID: 24113238
-
Computational methods in the discovery and design of BACE-1 inhibitors.Curr Med Chem. 2012;19(36):6095-111. Curr Med Chem. 2012. PMID: 23072352 Review.
-
Progress toward the discovery and development of efficacious BACE inhibitors.Curr Opin Drug Discov Devel. 2006 Nov;9(6):776-91. Curr Opin Drug Discov Devel. 2006. PMID: 17117686 Review.
Cited by
-
Spatiotemporal identification of druggable binding sites using deep learning.Commun Biol. 2020 Oct 27;3(1):618. doi: 10.1038/s42003-020-01350-0. Commun Biol. 2020. PMID: 33110179 Free PMC article.
-
Convolidine as potent BACE1 inhibitor for Alzheimer's disease; in-silico coupled with in-vitro assessment.J Comput Aided Mol Des. 2025 Apr 10;39(1):13. doi: 10.1007/s10822-025-00592-6. J Comput Aided Mol Des. 2025. PMID: 40208466
-
Computer-Aided Ligand Discovery for Estrogen Receptor Alpha.Int J Mol Sci. 2020 Jun 12;21(12):4193. doi: 10.3390/ijms21124193. Int J Mol Sci. 2020. PMID: 32545494 Free PMC article. Review.
-
Allosteric binding sites in Rab11 for potential drug candidates.PLoS One. 2018 Jun 6;13(6):e0198632. doi: 10.1371/journal.pone.0198632. eCollection 2018. PLoS One. 2018. PMID: 29874286 Free PMC article.
-
Acetylcholinesterase: A Versatile Template to Coin Potent Modulators of Multiple Therapeutic Targets.Acc Chem Res. 2024 Feb 9;57(4):450-67. doi: 10.1021/acs.accounts.3c00617. Online ahead of print. Acc Chem Res. 2024. PMID: 38333993 Free PMC article.
References
-
- Selkoe DJ. Translating cell biology into therapeutic in Alzheimer’s disease. Nature. 1999; 399:A23–31. - PubMed
-
- Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum Mol Genet. 2001; 10:1317–1324. - PubMed
