

Vipie: web pipeline for parallel characterization of viral populations from multiple NGS samples
- PMID: 28506246
- PMCID: PMC5430618
- DOI: 10.1186/s12864-017-3721-7
Vipie: web pipeline for parallel characterization of viral populations from multiple NGS samples
Abstract
Background: Next generation sequencing (NGS) technology allows laboratories to investigate virome composition in clinical and environmental samples in a culture-independent way. There is a need for bioinformatic tools capable of parallel processing of virome sequencing data by exactly identical methods: this is especially important in studies of multifactorial diseases, or in parallel comparison of laboratory protocols.
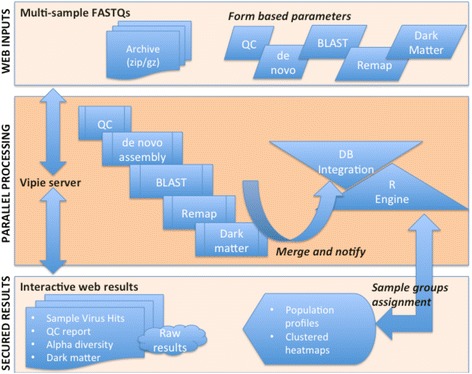
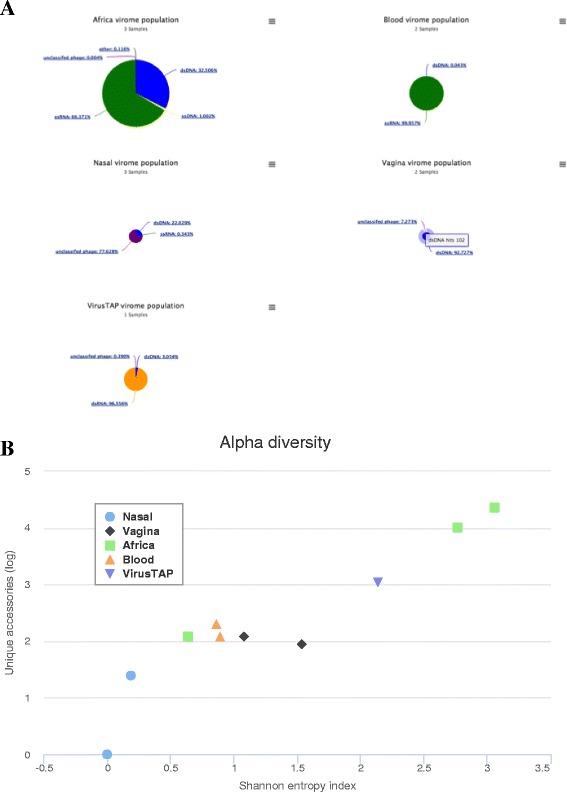
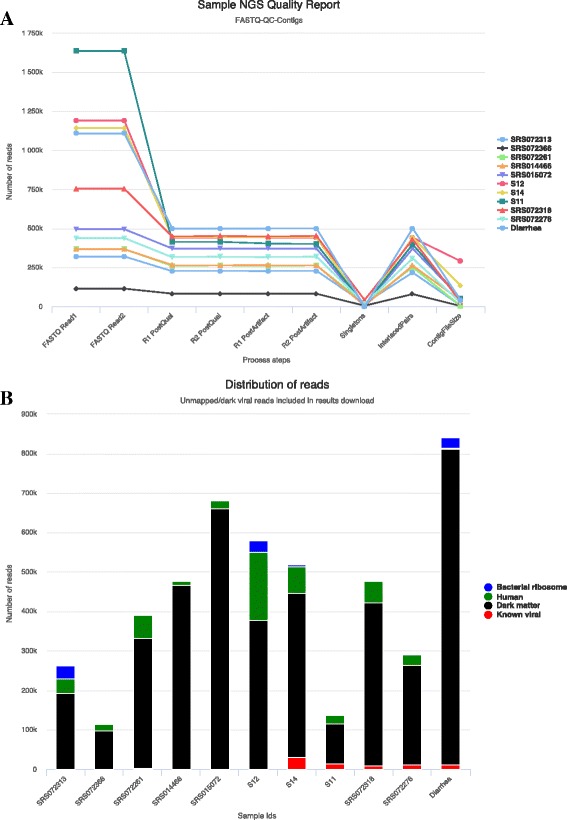
Results: We have developed a web-based application allowing direct upload of sequences from multiple virome samples using custom parameters. The samples are then processed in parallel using an identical protocol, and can be easily reanalyzed. The pipeline performs de-novo assembly, taxonomic classification of viruses as well as sample analyses based on user-defined grouping categories. Tables of virus abundance are produced from cross-validation by remapping the sequencing reads to a union of all observed reference viruses. In addition, read sets and reports are created after processing unmapped reads against known human and bacterial ribosome references. Secured interactive results are dynamically plotted with population and diversity charts, clustered heatmaps and a sortable and searchable abundance table.
Conclusions: The Vipie web application is a unique tool for multi-sample metagenomic analysis of viral data, producing searchable hits tables, interactive population maps, alpha diversity measures and clustered heatmaps that are grouped in applicable custom sample categories. Known references such as human genome and bacterial ribosomal genes are optionally removed from unmapped ('dark matter') reads. Secured results are accessible and shareable on modern browsers. Vipie is a freely available web-based tool whose code is open source.
Keywords: Assembly; Metagenomics; NGS analysis; Parallel processing; Viral dark matter; Viromes; Virus; Visualization.
Figures

Similar articles
-
ViraPipe: scalable parallel pipeline for viral metagenome analysis from next generation sequencing reads.Bioinformatics. 2018 Mar 15;34(6):928-935. doi: 10.1093/bioinformatics/btx702. Bioinformatics. 2018. PMID: 29106455
-
Improved Assembly of Metagenome-Assembled Genomes and Viruses in Tibetan Saline Lake Sediment by HiFi Metagenomic Sequencing.Microbiol Spectr. 2023 Feb 14;11(1):e0332822. doi: 10.1128/spectrum.03328-22. Epub 2022 Dec 8. Microbiol Spectr. 2023. PMID: 36475839 Free PMC article.
-
Virus detection in high-throughput sequencing data without a reference genome of the host.Infect Genet Evol. 2018 Dec;66:180-187. doi: 10.1016/j.meegid.2018.09.026. Epub 2018 Oct 3. Infect Genet Evol. 2018. PMID: 30292006
-
Recent advances in inferring viral diversity from high-throughput sequencing data.Virus Res. 2017 Jul 15;239:17-32. doi: 10.1016/j.virusres.2016.09.016. Epub 2016 Sep 28. Virus Res. 2017. PMID: 27693290 Review.
-
NGSPanPipe: A Pipeline for Pan-genome Identification in Microbial Strains from Experimental Reads.Adv Exp Med Biol. 2018;1052:39-49. doi: 10.1007/978-981-10-7572-8_4. Adv Exp Med Biol. 2018. PMID: 29785479 Review.
Cited by
-
Evaluating the Alimentary and Respiratory Tracts in Health and disease (EARTH) research programme: a protocol for prospective, longitudinal, controlled, observational studies in children with chronic disease at an Australian tertiary paediatric hospital.BMJ Open. 2020 Apr 14;10(4):e033916. doi: 10.1136/bmjopen-2019-033916. BMJ Open. 2020. PMID: 32295774 Free PMC article.
-
Metagenomics of the faecal virome indicate a cumulative effect of enterovirus and gluten amount on the risk of coeliac disease autoimmunity in genetically at risk children: the TEDDY study.Gut. 2020 Aug;69(8):1416-1422. doi: 10.1136/gutjnl-2019-319809. Epub 2019 Nov 19. Gut. 2020. PMID: 31744911 Free PMC article.
-
Next Generation Sequencing Approaches to Characterize the Respiratory Tract Virome.Microorganisms. 2022 Nov 24;10(12):2327. doi: 10.3390/microorganisms10122327. Microorganisms. 2022. PMID: 36557580 Free PMC article. Review.
-
HPV-EM: an accurate HPV detection and genotyping EM algorithm.Sci Rep. 2020 Aug 31;10(1):14340. doi: 10.1038/s41598-020-71300-7. Sci Rep. 2020. PMID: 32868873 Free PMC article.
-
VirID: Beyond Virus Discovery-An Integrated Platform for Comprehensive RNA Virus Characterization.Mol Biol Evol. 2024 Oct 4;41(10):msae202. doi: 10.1093/molbev/msae202. Mol Biol Evol. 2024. PMID: 39331699 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
