

Quantifying the mapping precision of genome-wide association studies using whole-genome sequencing data
- PMID: 28506277
- PMCID: PMC5432979
- DOI: 10.1186/s13059-017-1216-0
Quantifying the mapping precision of genome-wide association studies using whole-genome sequencing data
Abstract
Background: Understanding the mapping precision of genome-wide association studies (GWAS), that is the physical distances between the top associated single-nucleotide polymorphisms (SNPs) and the causal variants, is essential to design fine-mapping experiments for complex traits and diseases.
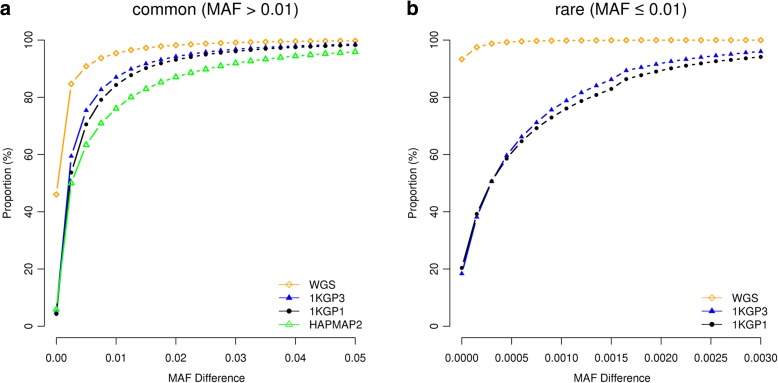
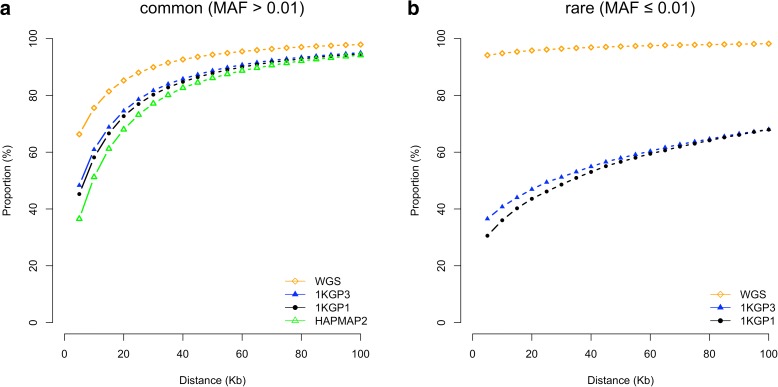
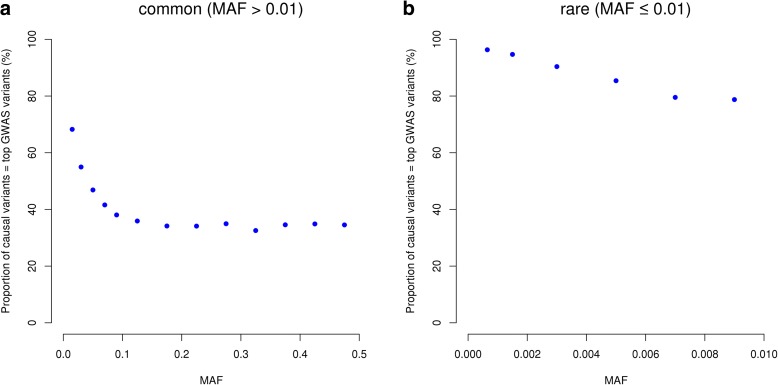
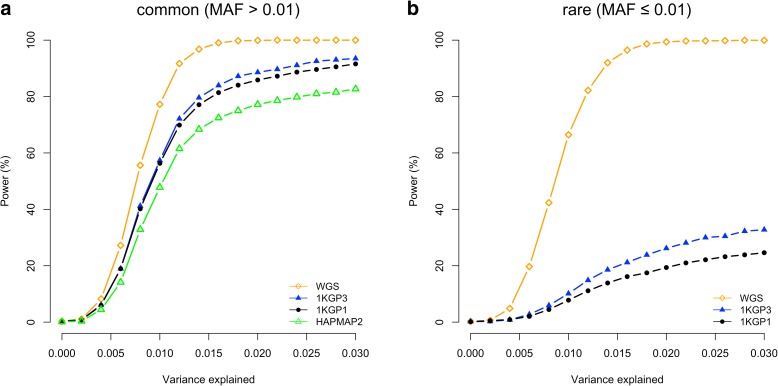
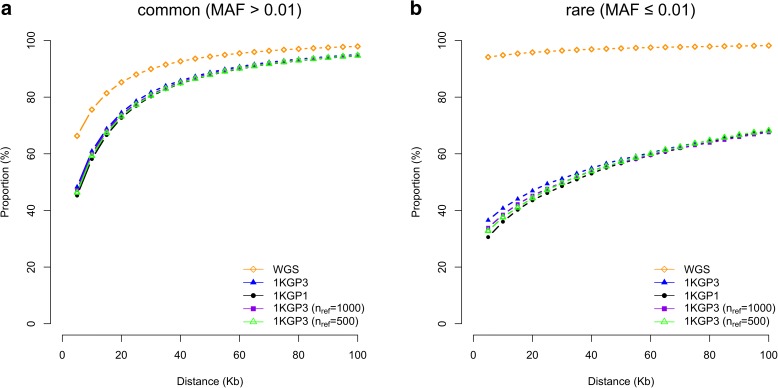
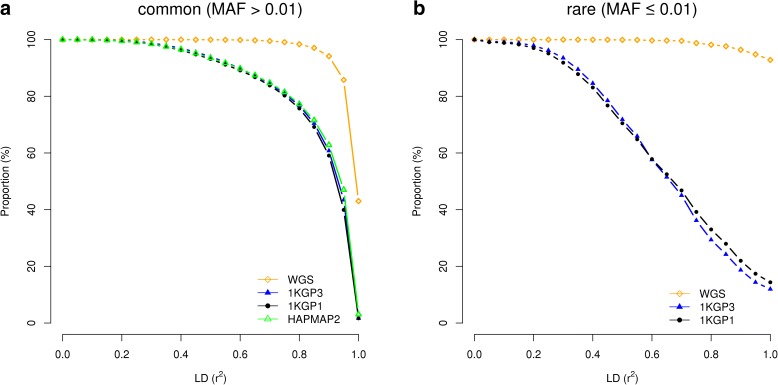
Results: Using simulations based on whole-genome sequencing (WGS) data from 3642 unrelated individuals of European descent, we show that the association signals at rare causal variants (minor allele frequency ≤ 0.01) are very unlikely to be mapped to common variants in GWAS using either WGS data or imputed data and vice versa. We predict that at least 80% of the common variants identified from published GWAS using imputed data are within 33.5 Kbp of the causal variants, a resolution that is comparable with that using WGS data. Mapping precision at these loci will improve with increasing sample sizes of GWAS in the future. For rare variants, the mapping precision of GWAS using WGS data is extremely high, suggesting WGS is an efficient strategy to detect and fine-map rare variants simultaneously. We further assess the mapping precision by linkage disequilibrium between GWAS hits and causal variants and develop an online tool (gwasMP) to query our results with different thresholds of physical distance and/or linkage disequilibrium ( http://cnsgenomics.com/shiny/gwasMP ).
Conclusions: Our findings provide a benchmark to inform future design and development of fine-mapping experiments and technologies to pinpoint the causal variants at GWAS loci.
Keywords: False positive rate; Genome-wide association studies; Imputation; Mapping precision; Whole genome sequencing.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
