

ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost
- PMID: 28507695
- PMCID: PMC5414547
- DOI: 10.1039/c6sc05720a
ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost
Abstract
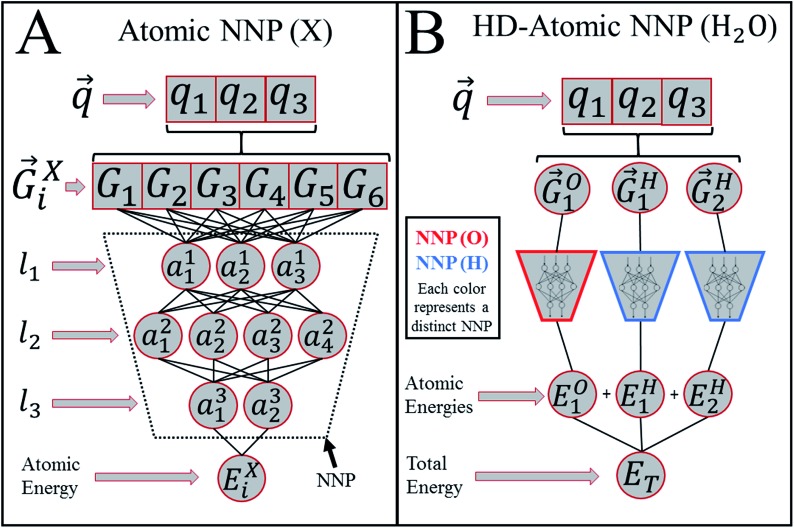
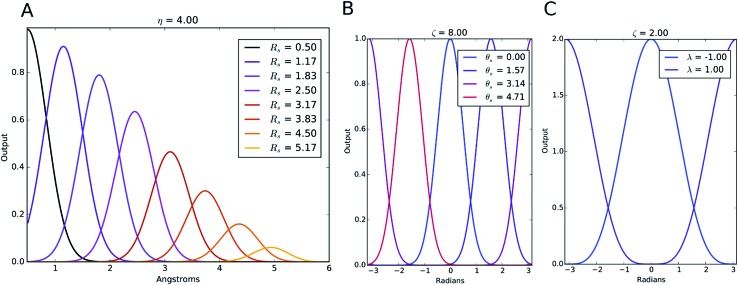
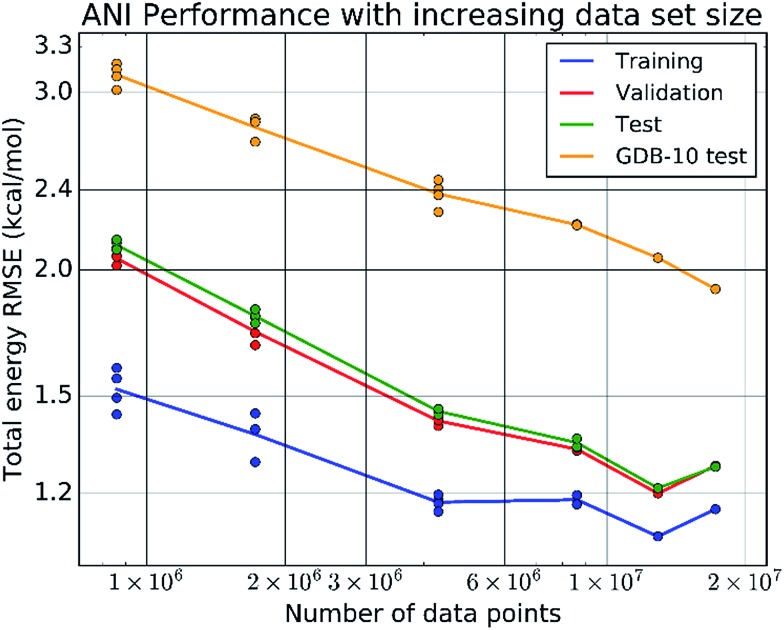
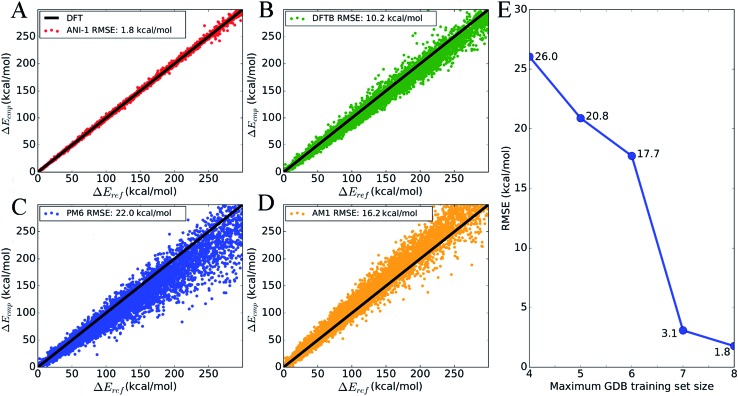
Deep learning is revolutionizing many areas of science and technology, especially image, text, and speech recognition. In this paper, we demonstrate how a deep neural network (NN) trained on quantum mechanical (QM) DFT calculations can learn an accurate and transferable potential for organic molecules. We introduce ANAKIN-ME (Accurate NeurAl networK engINe for Molecular Energies) or ANI for short. ANI is a new method designed with the intent of developing transferable neural network potentials that utilize a highly-modified version of the Behler and Parrinello symmetry functions to build single-atom atomic environment vectors (AEV) as a molecular representation. AEVs provide the ability to train neural networks to data that spans both configurational and conformational space, a feat not previously accomplished on this scale. We utilized ANI to build a potential called ANI-1, which was trained on a subset of the GDB databases with up to 8 heavy atoms in order to predict total energies for organic molecules containing four atom types: H, C, N, and O. To obtain an accelerated but physically relevant sampling of molecular potential surfaces, we also proposed a Normal Mode Sampling (NMS) method for generating molecular conformations. Through a series of case studies, we show that ANI-1 is chemically accurate compared to reference DFT calculations on much larger molecular systems (up to 54 atoms) than those included in the training data set.
Figures
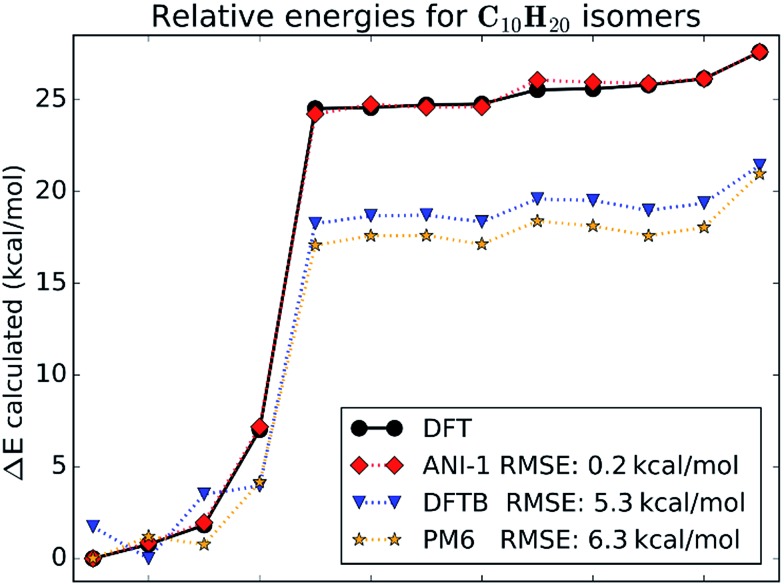
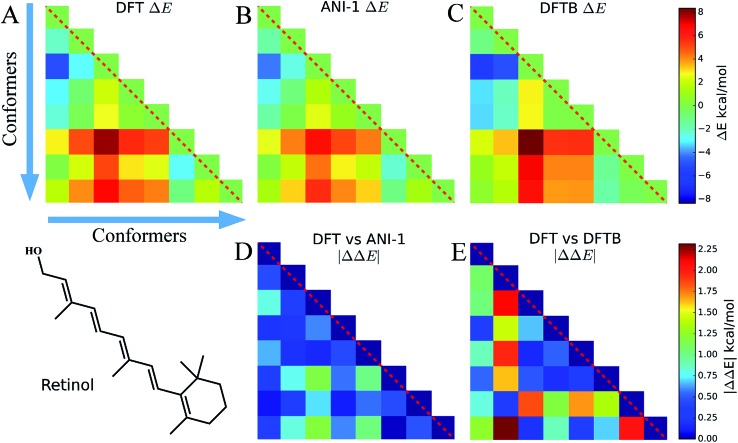
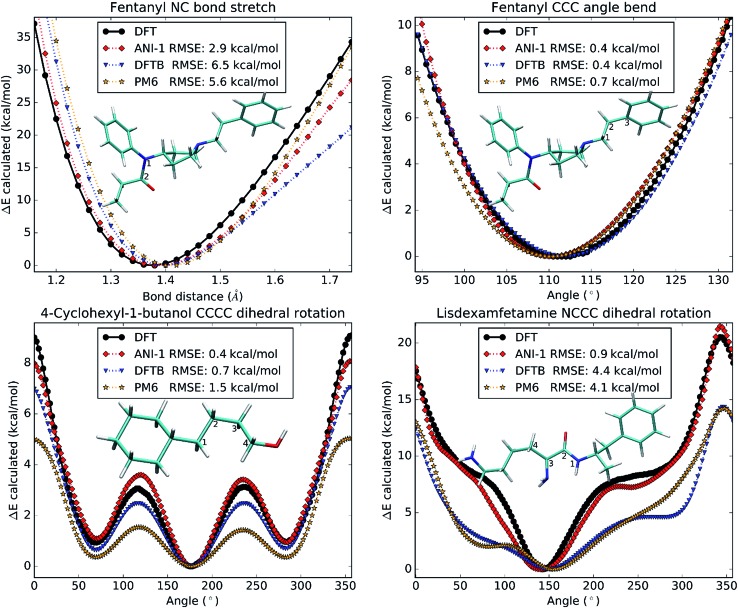
References
-
- Kitaura K., Ikeo E., Asada T., Nakano T., Uebayasi M. Chem. Phys. Lett. 1999;313:701–706.
-
- Fedorov D. G., Nagata T., Kitaura K. Phys. Chem. Chem. Phys. 2012;14:7562. - PubMed
-
- Ochsenfeld C., Kussmann J. and Lambrecht D. S., in Reviews in Computational Chemistry, John Wiley & Sons, Inc., 2007, pp. 1–82.
-
- Elstner M. Theor. Chem. Acc. 2006;116:316–325.
-
- Stewart J. J. P. J. Mol. Model. 2009;15:765–805. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources