

Machine learning of accurate energy-conserving molecular force fields
- PMID: 28508076
- PMCID: PMC5419702
- DOI: 10.1126/sciadv.1603015
Machine learning of accurate energy-conserving molecular force fields
Abstract
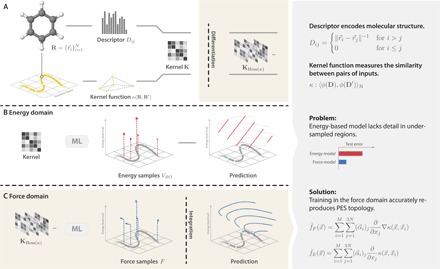
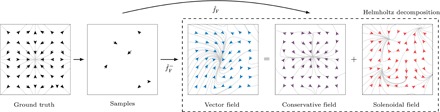
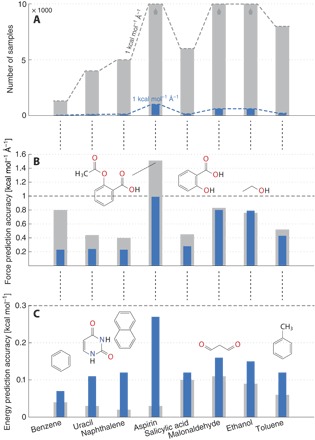
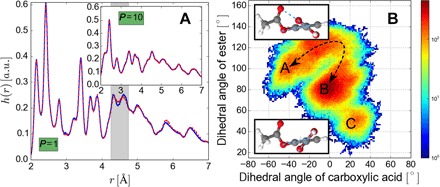
Using conservation of energy-a fundamental property of closed classical and quantum mechanical systems-we develop an efficient gradient-domain machine learning (GDML) approach to construct accurate molecular force fields using a restricted number of samples from ab initio molecular dynamics (AIMD) trajectories. The GDML implementation is able to reproduce global potential energy surfaces of intermediate-sized molecules with an accuracy of 0.3 kcal mol-1 for energies and 1 kcal mol-1 Å̊-1 for atomic forces using only 1000 conformational geometries for training. We demonstrate this accuracy for AIMD trajectories of molecules, including benzene, toluene, naphthalene, ethanol, uracil, and aspirin. The challenge of constructing conservative force fields is accomplished in our work by learning in a Hilbert space of vector-valued functions that obey the law of energy conservation. The GDML approach enables quantitative molecular dynamics simulations for molecules at a fraction of cost of explicit AIMD calculations, thereby allowing the construction of efficient force fields with the accuracy and transferability of high-level ab initio methods.
Keywords: energy conservation; force field; gradient field; kernel regression; machine learning; molecular dynamics; path integrals; potential-energy surface.
Figures
References
-
- Behler J., Parrinello M., Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007). - PubMed
-
- Behler J., Lorenz S., Reuter K., Representing molecule-surface interactions with symmetry-adapted neural networks. J. Chem. Phys. 127, 014705 (2007). - PubMed
-
- Bartók A. P., Payne M. C., Kondor R., Csányi G., Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 104, 136403 (2010). - PubMed
-
- Behler J., Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys. 134, 074106 (2011). - PubMed
-
- Behler J., Neural network potential-energy surfaces in chemistry: A tool for large-scale simulations. Phys. Chem. Chem. Phys. 13, 17930–17955 (2011). - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
