

Clinicopathologic and molecular spectrum of RNASEH1-related mitochondrial disease
- PMID: 28508084
- PMCID: PMC5413961
- DOI: 10.1212/NXG.0000000000000149
Clinicopathologic and molecular spectrum of RNASEH1-related mitochondrial disease
Abstract
Objective: Pathologic ribonuclease H1 (RNase H1) causes aberrant mitochondrial DNA (mtDNA) segregation and is associated with multiple mtDNA deletions. We aimed to determine the prevalence of RNase H1 gene (RNASEH1) mutations among patients with mitochondrial disease and establish clinically meaningful genotype-phenotype correlations.
Methods: RNASEH1 was analyzed in patients with (1) multiple deletions/depletion of muscle mtDNA and (2) mendelian progressive external ophthalmoplegia (PEO) with neuropathologic evidence of mitochondrial dysfunction, but no detectable multiple deletions/depletion of muscle mtDNA. Clinicopathologic and molecular evaluation of the newly identified and previously reported patients harboring RNASEH1 mutations was subsequently undertaken.
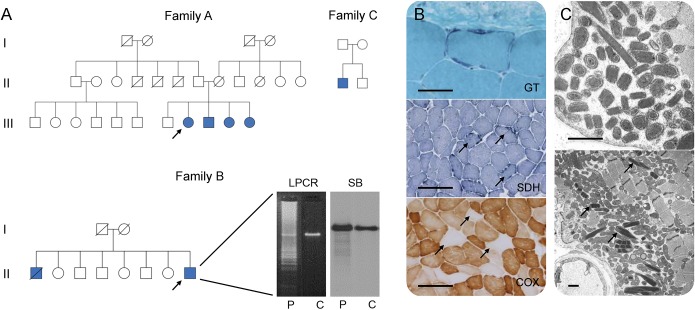
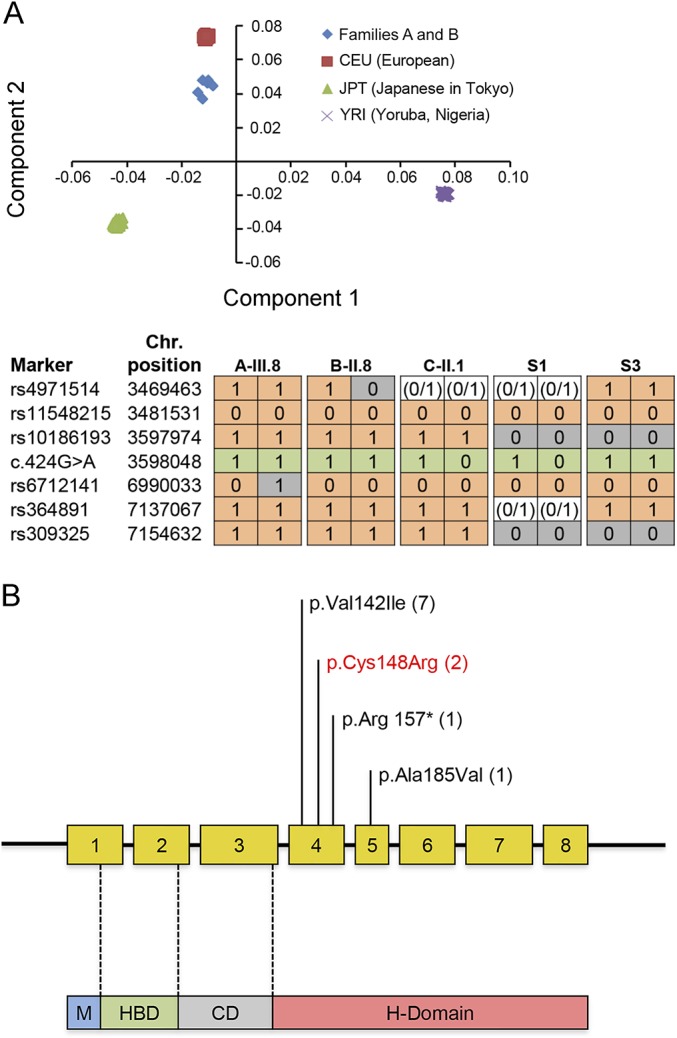
Results: Pathogenic c.424G>A p.Val142Ile RNASEH1 mutations were detected in 3 pedigrees among the 74 probands screened. Given that all 3 families had Indian ancestry, RNASEH1 genetic analysis was undertaken in 50 additional Indian probands with variable clinical presentations associated with multiple mtDNA deletions, but no further RNASEH1 mutations were confirmed. RNASEH1-related mitochondrial disease was characterized by PEO (100%), cerebellar ataxia (57%), and dysphagia (50%). The ataxia neuropathy spectrum phenotype was observed in 1 patient. Although the c.424G>A p.Val142Ile mutation underpins all reported RNASEH1-related mitochondrial disease, haplotype analysis suggested an independent origin, rather than a founder event, for the variant in our families.
Conclusions: In our cohort, RNASEH1 mutations represent the fourth most common cause of adult mendelian PEO associated with multiple mtDNA deletions, following mutations in POLG, RRM2B, and TWNK. RNASEH1 genetic analysis should also be considered in all patients with POLG-negative ataxia neuropathy spectrum. The pathophysiologic mechanisms by which the c.424G>A p.Val142Ile mutation impairs human RNase H1 warrant further investigation.
Figures
References
-
- Cerritelli SM, Frolova EG, Feng C, Grinberg A, Love PE, Crouch RJ. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol Cell 2003;11:807–815. - PubMed
Grants and funding
- MR/K000608/1/MRC_/Medical Research Council/United Kingdom
- G1001253/MRC_/Medical Research Council/United Kingdom
- G108/638/MRC_/Medical Research Council/United Kingdom
- MC_PC_13029/MRC_/Medical Research Council/United Kingdom
- MR/J010448/1/MRC_/Medical Research Council/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- MC_PC_13029/2/MRC_/Medical Research Council/United Kingdom
- G0601943/MRC_/Medical Research Council/United Kingdom
- G0800674/MRC_/Medical Research Council/United Kingdom
- MC_UP_1002/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00015/5/MRC_/Medical Research Council/United Kingdom
- MC_U105663140/MRC_/Medical Research Council/United Kingdom
- MR/J004758/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00015/8/MRC_/Medical Research Council/United Kingdom
- MC_UP_1202/14/MRC_/Medical Research Council/United Kingdom
- G0802760/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources