

Receptor-ligand molecular docking
- PMID: 28509958
- PMCID: PMC5425711
- DOI: 10.1007/s12551-013-0130-2
Receptor-ligand molecular docking
Abstract
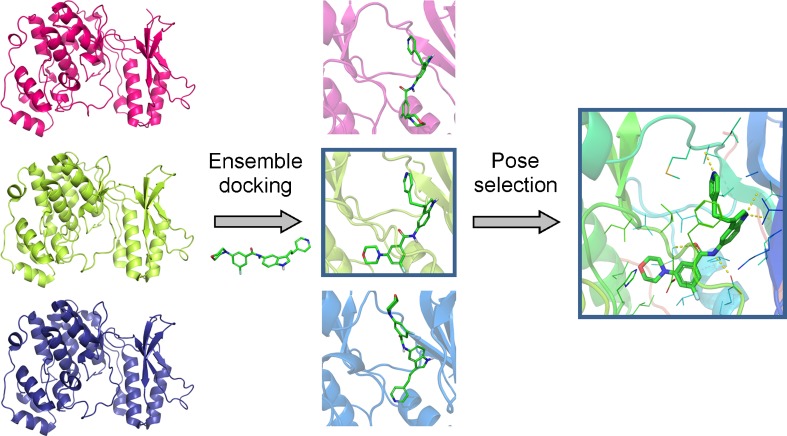
Docking methodology aims to predict the experimental binding modes and affinities of small molecules within the binding site of particular receptor targets and is currently used as a standard computational tool in drug design for lead compound optimisation and in virtual screening studies to find novel biologically active molecules. The basic tools of a docking methodology include a search algorithm and an energy scoring function for generating and evaluating ligand poses. In this review, we present the search algorithms and scoring functions most commonly used in current molecular docking methods that focus on protein-ligand applications. We summarise the main topics and recent computational and methodological advances in protein-ligand docking. Protein flexibility, multiple ligand binding modes and the free-energy landscape profile for binding affinity prediction are important and interconnected challenges to be overcome by further methodological developments in the docking field.
Keywords: Protein-ligand docking; Scoring functions; Search algorithms; Structure-based drug design.
Figures
References
-
- Abagyan R, Totrov M, Kuznetsov D. ICM: A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15:488–506.
-
- Apostolakis J, Plückthun A, Caflisch A. Docking small ligands in flexible binding sites. J Comput Chem. 1998;19:21–37.
-
- Aqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994;7:385–391. - PubMed
-
- Asses Y, Venkatraman V, Leroux V, et al. Exploring c-Met kinase flexibility by sampling and clustering its conformational space. Proteins Struct Funct Bioinform. 2012;80:1227–1238. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
