

nNOS regulation of skeletal muscle fatigue and exercise performance
- PMID: 28510048
- PMCID: PMC5425689
- DOI: 10.1007/s12551-011-0060-9
nNOS regulation of skeletal muscle fatigue and exercise performance
Abstract
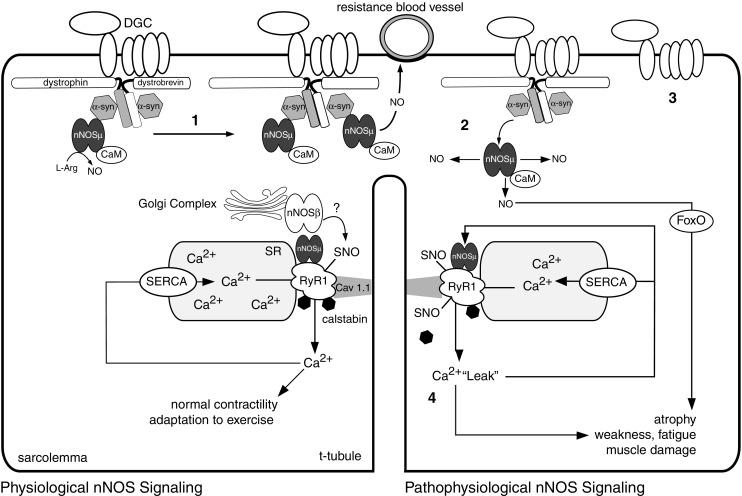
Neuronal nitric oxide synthases (nNOS) are Ca2+/calmodulin-activated enzymes that synthesize the gaseous messenger nitric oxide (NO). nNOSμ and the recently described nNOSβ, both spliced nNOS isoforms, are important enzymatic sources of NO in skeletal muscle, a tissue long considered to be a paradigmatic system for studying NO-dependent redox signaling. nNOS is indispensable for skeletal muscle integrity and contractile performance, and deregulation of nNOSμ signaling is a common pathogenic feature of many neuromuscular diseases. Recent evidence suggests that both nNOSμ and nNOSβ regulate skeletal muscle size, strength, and fatigue resistance, making them important players in exercise performance. nNOSμ acts as an activity sensor and appears to assist skeletal muscle adaptation to new functional demands, particularly those of endurance exercise. Prolonged inactivity leads to nNOS-mediated muscle atrophy through a FoxO-dependent pathway. nNOS also plays a role in modulating exercise performance in neuromuscular disease. In the mdx mouse model of Duchenne muscular dystrophy, defective nNOS signaling is thought to restrict contractile capacity of working muscle in two ways: loss of sarcolemmal nNOSμ causes excessive ischemic damage while residual cytosolic nNOSμ contributes to hypernitrosylation of the ryanodine receptor, causing pathogenic Ca2+ leak. This defect in Ca2+ handling promotes muscle damage, weakness, and fatigue. This review addresses these recent advances in the understanding of nNOS-dependent redox regulation of skeletal muscle function and exercise performance under physiological and neuromuscular disease conditions.
Keywords: Dystrophin; Fatigue; Nitric oxide; Nitrosylation; Ryanodine receptor; nNOS.
Figures
Similar articles
-
Loss of nNOS inhibits compensatory muscle hypertrophy and exacerbates inflammation and eccentric contraction-induced damage in mdx mice.Hum Mol Genet. 2015 Jan 15;24(2):492-505. doi: 10.1093/hmg/ddu469. Epub 2014 Sep 11. Hum Mol Genet. 2015. PMID: 25214536 Free PMC article.
-
Golgi and sarcolemmal neuronal NOS differentially regulate contraction-induced fatigue and vasoconstriction in exercising mouse skeletal muscle.J Clin Invest. 2010 Mar;120(3):816-26. doi: 10.1172/JCI40736. J Clin Invest. 2010. PMID: 20124730 Free PMC article.
-
Functional deficits in nNOSmu-deficient skeletal muscle: myopathy in nNOS knockout mice.PLoS One. 2008;3(10):e3387. doi: 10.1371/journal.pone.0003387. Epub 2008 Oct 13. PLoS One. 2008. PMID: 18852886 Free PMC article.
-
Neuronal nitric oxide synthase (nNOS) splice variant function: Insights into nitric oxide signaling from skeletal muscle.Nitric Oxide. 2019 Jan 1;82:35-47. doi: 10.1016/j.niox.2018.11.004. Epub 2018 Nov 29. Nitric Oxide. 2019. PMID: 30503614 Review.
-
Nitric Oxide Regulates Skeletal Muscle Fatigue, Fiber Type, Microtubule Organization, and Mitochondrial ATP Synthesis Efficiency Through cGMP-Dependent Mechanisms.Antioxid Redox Signal. 2017 Jun 10;26(17):966-985. doi: 10.1089/ars.2016.6630. Epub 2016 Aug 17. Antioxid Redox Signal. 2017. PMID: 27393340 Free PMC article. Review.
Cited by
-
Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy.J Pathol. 2012 Sep;228(1):77-87. doi: 10.1002/path.4054. Epub 2012 Jul 18. J Pathol. 2012. PMID: 22653783 Free PMC article.
-
nNOS/GSNOR interaction contributes to skeletal muscle differentiation and homeostasis.Cell Death Dis. 2019 May 1;10(5):354. doi: 10.1038/s41419-019-1584-3. Cell Death Dis. 2019. PMID: 31043586 Free PMC article.
-
Loss of nNOS inhibits compensatory muscle hypertrophy and exacerbates inflammation and eccentric contraction-induced damage in mdx mice.Hum Mol Genet. 2015 Jan 15;24(2):492-505. doi: 10.1093/hmg/ddu469. Epub 2014 Sep 11. Hum Mol Genet. 2015. PMID: 25214536 Free PMC article.
-
The Molecular Simulation Study of nNOS Activation Induced by the Interaction Between Its Calmodulin-Binding Domain and SUMO1.Front Mol Neurosci. 2020 Oct 29;13:535494. doi: 10.3389/fnmol.2020.535494. eCollection 2020. Front Mol Neurosci. 2020. PMID: 33192289 Free PMC article.
-
Lactobacillus fermentum CQPC08 Attenuates Exercise-Induced Fatigue in Mice Through Its Antioxidant Effects and Effective Intervention of Galactooligosaccharide.Drug Des Devel Ther. 2021 Dec 24;15:5151-5164. doi: 10.2147/DDDT.S317456. eCollection 2021. Drug Des Devel Ther. 2021. PMID: 34992351 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
