

Toward an elucidation of the molecular genetics of inherited retinal degenerations
- PMID: 28510639
- PMCID: PMC5886474
- DOI: 10.1093/hmg/ddx185
Toward an elucidation of the molecular genetics of inherited retinal degenerations
Abstract
While individually classed as rare diseases, hereditary retinal degenerations (IRDs) are the major cause of registered visual handicap in the developed world. Given their hereditary nature, some degree of intergenic heterogeneity was expected, with genes segregating in autosomal dominant, recessive, X-linked recessive, and more rarely in digenic or mitochondrial modes. Today, it is recognized that IRDs, as a group, represent one of the most genetically diverse of hereditary conditions - at least 260 genes having been implicated, with 70 genes identified in the most common IRD, retinitis pigmentosa (RP). However, targeted sequencing studies of exons from known IRD genes have resulted in the identification of candidate mutations in only approximately 60% of IRD cases. Given recent advances in the development of gene-based medicines, characterization of IRD patient cohorts for known IRD genes and elucidation of the molecular pathologies of disease in those remaining unresolved cases has become an endeavor of the highest priority. Here, we provide an outline of progress in this area.
© The Author 2017. Published by Oxford University Press.
Figures
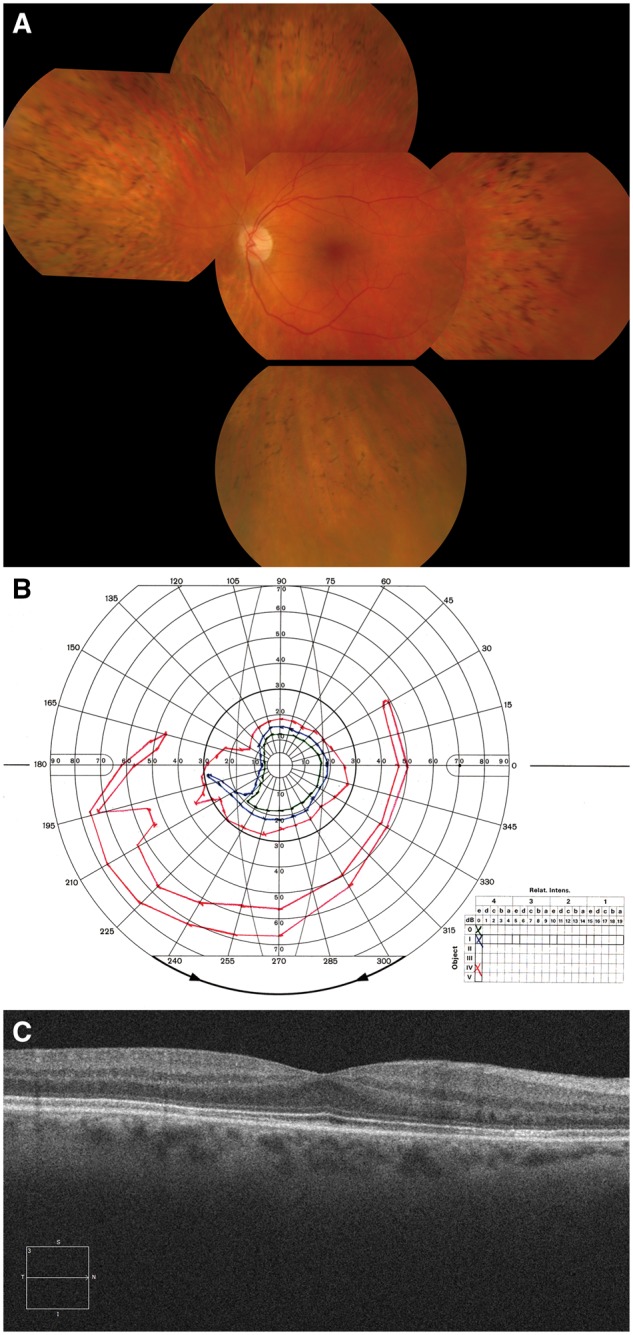
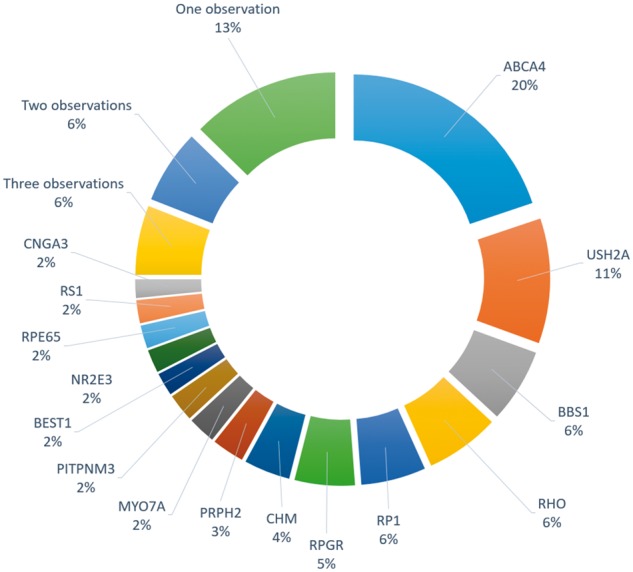
References
-
- Bhattacharya S.S., Wright A.F., Clayton J.F., Price W.H., Phillips C.I., McKeown C.M., Jay M., Bird A.C., Pearson P.L., Southern E.M.. et al. (1984) Close genetic linkage between X-linked retinitis pigmentosa and a restriction fragment length polymorphism identified by recombinant DNA probe L1.28. Nature, 309, 253–255. - PubMed
-
- McWilliam P., Farrar G.J., Kenna P.F., Bradley D.G., Humphries M.M., Sharp E.M., McConnell D.J., Lawler M., Sheils D., Ryan C., Humphries P. (1989) Genomics, 3, 619–622. - PubMed
-
- Farrar G.J., Kenna P.F., Jordan S.A., Kumar-Singh R., Humphries M.M., Sharp E.M., Sheils D.M., Humphries P. (1991) A three-base-pair deletion in the peripherin-RDS gene in one form of retinitis pigmentosa. Nature, 354, 478–480. - PubMed
-
- Chiang J.P., Trzupek K. (2015) The current status of molecular diagnosis of inherited retinal dystrophies. Curr. Opin. Ophthalmol., 26, 346–351. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
