

Variant effect prediction tools assessed using independent, functional assay-based datasets: implications for discovery and diagnostics
- PMID: 28511696
- PMCID: PMC5433009
- DOI: 10.1186/s40246-017-0104-8
Variant effect prediction tools assessed using independent, functional assay-based datasets: implications for discovery and diagnostics
Abstract
Background: Genetic variant effect prediction algorithms are used extensively in clinical genomics and research to determine the likely consequences of amino acid substitutions on protein function. It is vital that we better understand their accuracies and limitations because published performance metrics are confounded by serious problems of circularity and error propagation. Here, we derive three independent, functionally determined human mutation datasets, UniFun, BRCA1-DMS and TP53-TA, and employ them, alongside previously described datasets, to assess the pre-eminent variant effect prediction tools.
Results: Apparent accuracies of variant effect prediction tools were influenced significantly by the benchmarking dataset. Benchmarking with the assay-determined datasets UniFun and BRCA1-DMS yielded areas under the receiver operating characteristic curves in the modest ranges of 0.52 to 0.63 and 0.54 to 0.75, respectively, considerably lower than observed for other, potentially more conflicted datasets.
Conclusions: These results raise concerns about how such algorithms should be employed, particularly in a clinical setting. Contemporary variant effect prediction tools are unlikely to be as accurate at the general prediction of functional impacts on proteins as reported prior. Use of functional assay-based datasets that avoid prior dependencies promises to be valuable for the ongoing development and accurate benchmarking of such tools.
Keywords: Benchmarking; Functional assays; Functional datasets; Genomic screening; Mutation assessment; Pathogenicity prediction; Protein function; Variant effect prediction.
Figures
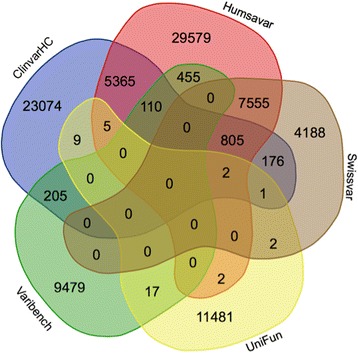
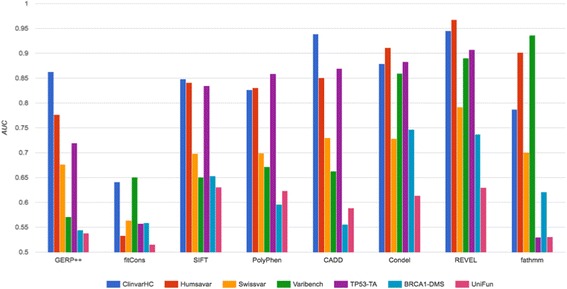
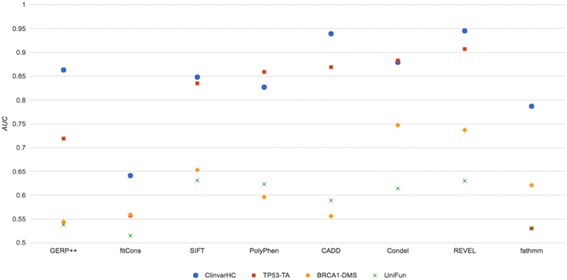
References
-
- Spurdle AB, Healey S, Devereau A, Hogervorst FBL, Monteiro ANA, Nathanson KL, et al. ENIGMA—evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat. 2012;33:2–7. doi: 10.1002/humu.21628. - DOI - PMC - PubMed
-
- Thompson BA, Spurdle AB, Plazzer J-P, Greenblatt MS, Akagi K, Al-Mulla F, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet Nature Research. 2013;46:107–115. doi: 10.1038/ng.2854. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
