

Structure-Based Mutations in the Herpes Simplex Virus 1 Glycoprotein B Ectodomain Arm Impart a Slow-Entry Phenotype
- PMID: 28512095
- PMCID: PMC5433099
- DOI: 10.1128/mBio.00614-17
Structure-Based Mutations in the Herpes Simplex Virus 1 Glycoprotein B Ectodomain Arm Impart a Slow-Entry Phenotype
Abstract
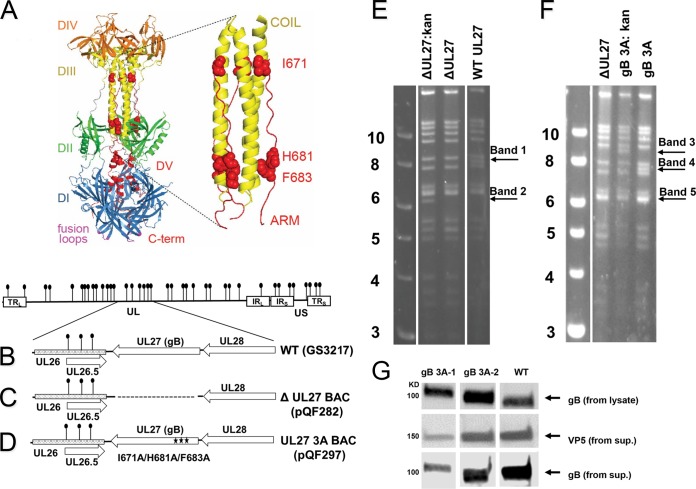
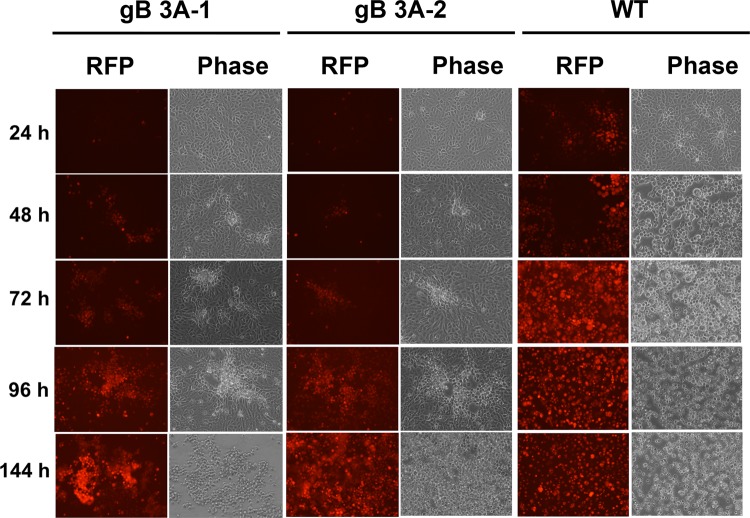
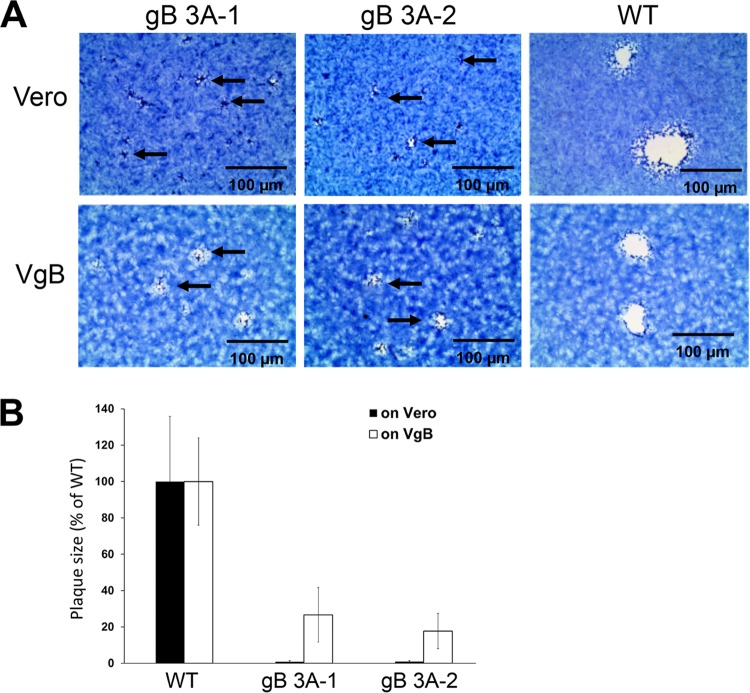
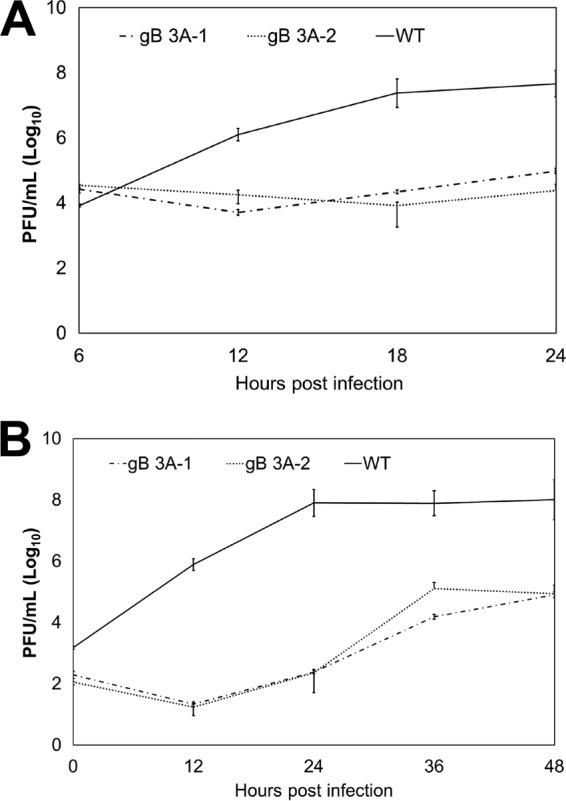
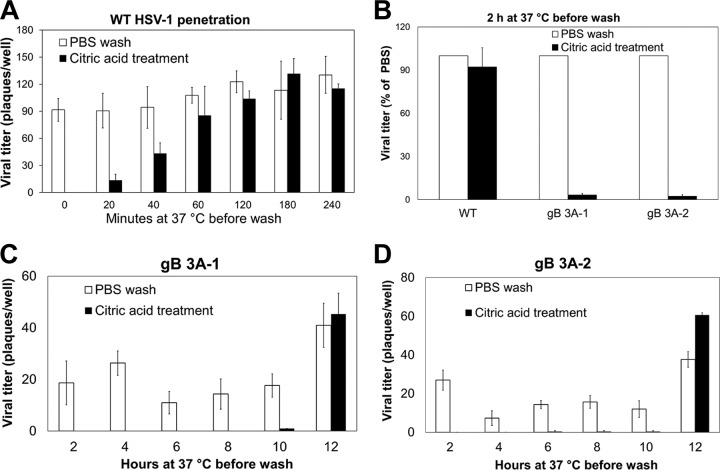
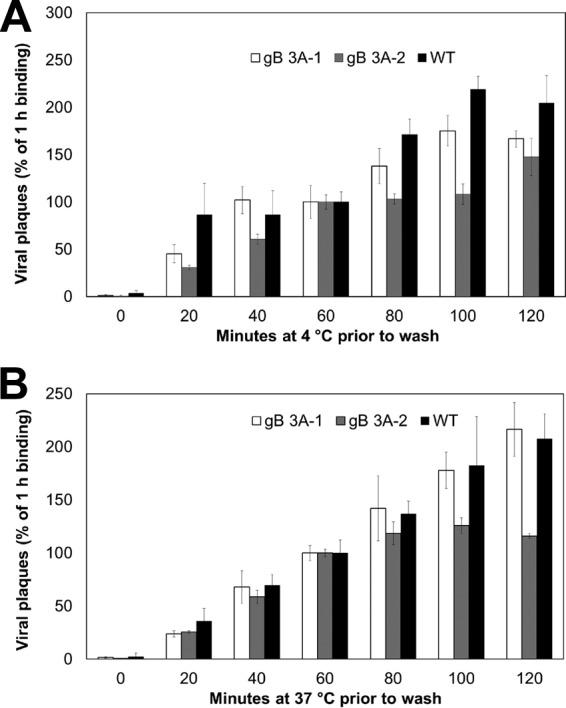
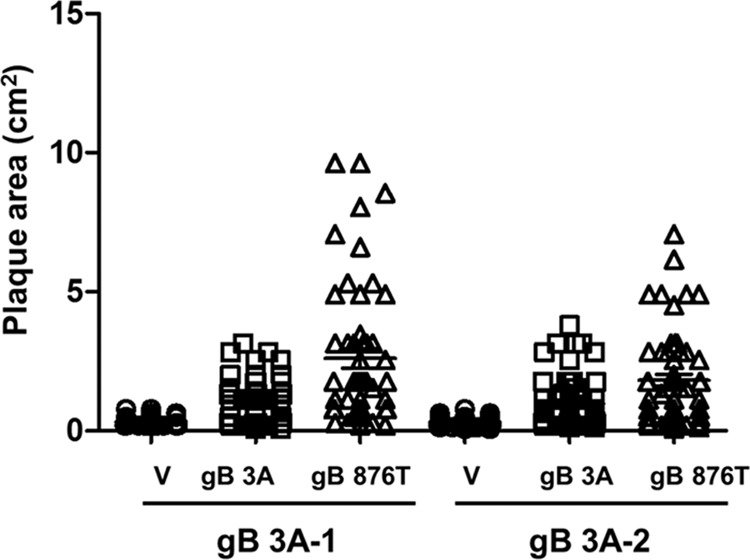
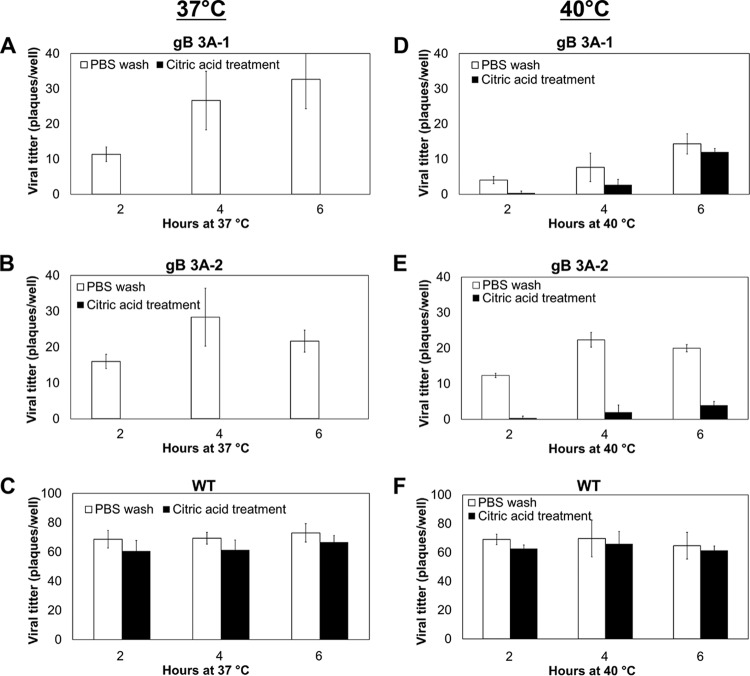
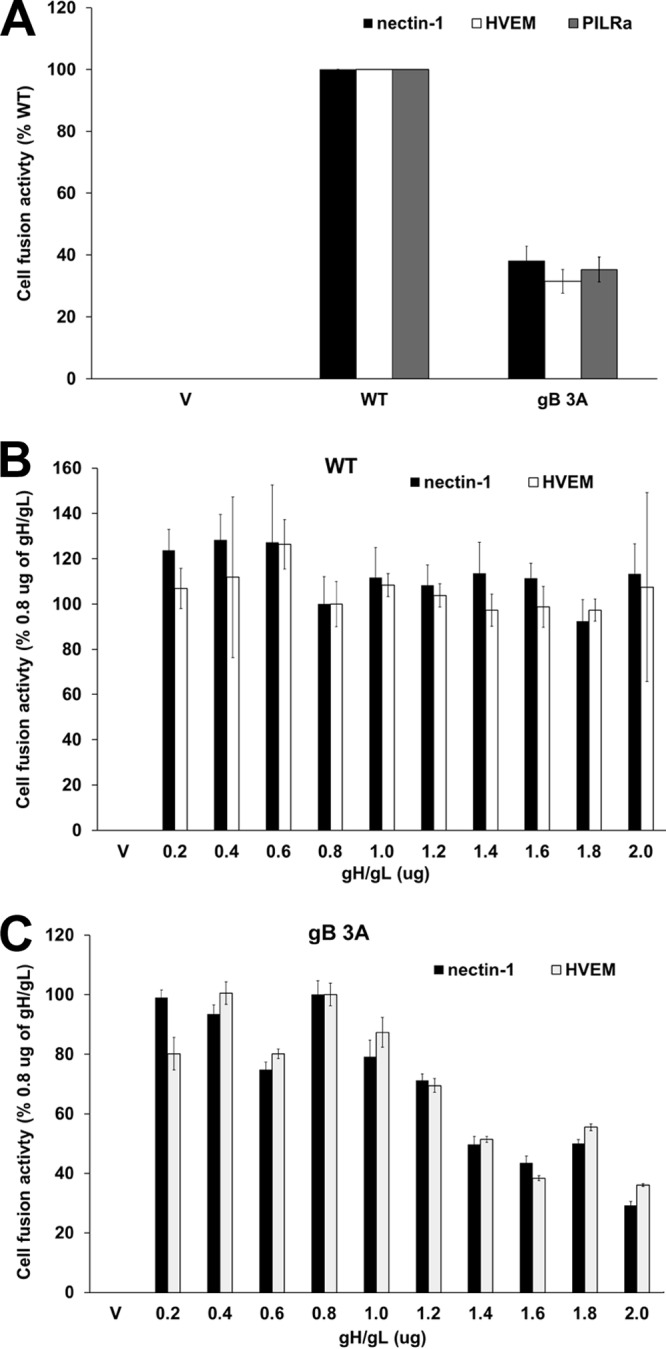
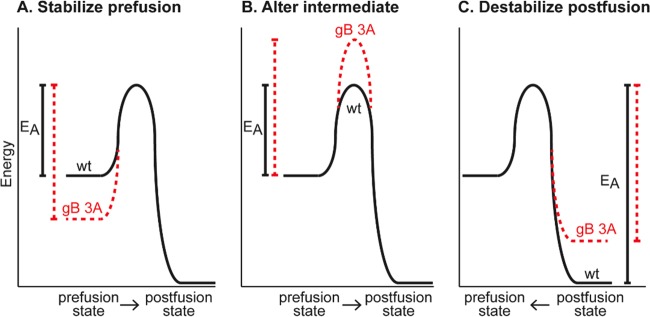
Glycoprotein B (gB) is the conserved herpesvirus fusion protein, and it is required for the entry of herpesviruses. The structure of the postfusion conformation of gB has been solved for several herpesviruses; however, the gB prefusion crystal structure and the details of how the protein refolds from a prefusion to a postfusion form to mediate fusion have not been determined. Using structure-based mutagenesis, we previously reported that three mutations (I671A, H681A, and F683A) in the C-terminal arm of the gB ectodomain greatly reduced cell-cell fusion. This fusion deficit could be rescued by the addition of a hyperfusogenic mutation, suggesting that the gB triple mutant was not misfolded. Using a bacterial artificial chromosome (BAC), we constructed two independent herpes simplex virus 1 mutant strains (gB 3A) carrying the three arm mutations. The gB 3A viruses have 200-fold smaller plaques than the wild-type virus and demonstrate remarkably delayed entry into cells. Single-step and multistep growth curves show that gB 3A viruses have delayed replication kinetics. Interestingly, incubation at 40°C promoted the entry of the gB 3A viruses. We propose that the gB 3A viruses' entry deficit is due to a loss of interactions between residues in the gB C-terminal arm and the coiled-coil core of gB. The results suggest that the triple alanine mutation may destabilize the postfusion gB conformation and/or stabilize the prefusion gB conformation and that exposure to elevated temperatures can overcome the defect in gB 3A viruses.IMPORTANCE Because of its complexity, the mechanism of herpesvirus entry into cells is not well understood. Our study investigated one of the most important unanswered questions about herpesvirus entry; i.e., how does the herpesvirus fusion protein gB mediate membrane fusion? gB is an essential protein that is conserved in all herpesviruses and is thought to undergo a conformational change to provide the energy to fuse the viral and cellular membranes. Using our understanding of the structure of gB, we designed mutations in the gB "arm" region that we predicted would impede gB function. We introduced these mutations into herpes simplex virus 1 by using a bacterial artificial chromosome, and the mutant virus exhibited a drastically delayed rate of entry. This entry defect was rescued by incubation at elevated temperatures, supporting a hypothesis that the engineered mutations altered the energetics of gB refolding. This study supports our hypothesis that an interaction between the gB arm and the core of gB is critical for gB refolding and the execution of membrane fusion, thus providing key details about the function of gB in herpesvirus-mediated fusion and subsequent virus entry.
Copyright © 2017 Fan et al.
Figures
Similar articles
-
Natural Selection of Glycoprotein B Mutations That Rescue the Small-Plaque Phenotype of a Fusion-Impaired Herpes Simplex Virus Mutant.mBio. 2018 Oct 16;9(5):e01948-18. doi: 10.1128/mBio.01948-18. mBio. 2018. PMID: 30327436 Free PMC article.
-
Residues within the C-terminal arm of the herpes simplex virus 1 glycoprotein B ectodomain contribute to its refolding during the fusion step of virus entry.J Virol. 2012 Jun;86(12):6386-93. doi: 10.1128/JVI.00104-12. Epub 2012 Apr 4. J Virol. 2012. PMID: 22491468 Free PMC article.
-
Herpes Simplex Virus Glycoprotein B Mutations Define Structural Sites in Domain I, the Membrane Proximal Region, and the Cytodomain That Regulate Entry.J Virol. 2021 Oct 27;95(22):e0105021. doi: 10.1128/JVI.01050-21. Epub 2021 Aug 25. J Virol. 2021. PMID: 34431697 Free PMC article.
-
Two Sides to Every Story: Herpes Simplex Type-1 Viral Glycoproteins gB, gD, gH/gL, gK, and Cellular Receptors Function as Key Players in Membrane Fusion.Viruses. 2021 Sep 16;13(9):1849. doi: 10.3390/v13091849. Viruses. 2021. PMID: 34578430 Free PMC article. Review.
-
The COMPLEXity in herpesvirus entry.Curr Opin Virol. 2017 Jun;24:97-104. doi: 10.1016/j.coviro.2017.04.006. Epub 2017 May 21. Curr Opin Virol. 2017. PMID: 28538165 Free PMC article. Review.
Cited by
-
Antiviral Activity of trans-Hexenoic Acid against Coxsackievirus B and Enterovirus A71.Antimicrob Agents Chemother. 2023 Mar 16;67(3):e0086822. doi: 10.1128/aac.00868-22. Epub 2023 Feb 14. Antimicrob Agents Chemother. 2023. PMID: 36786598 Free PMC article.
-
Integrin-Linked Kinase Reduces H3K9 Trimethylation to Enhance Herpes Simplex Virus 1 Replication.Front Cell Infect Microbiol. 2022 Mar 8;12:814307. doi: 10.3389/fcimb.2022.814307. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 35350437 Free PMC article.
-
Natural Selection of Glycoprotein B Mutations That Rescue the Small-Plaque Phenotype of a Fusion-Impaired Herpes Simplex Virus Mutant.mBio. 2018 Oct 16;9(5):e01948-18. doi: 10.1128/mBio.01948-18. mBio. 2018. PMID: 30327436 Free PMC article.
-
The structural basis of herpesvirus entry.Nat Rev Microbiol. 2021 Feb;19(2):110-121. doi: 10.1038/s41579-020-00448-w. Epub 2020 Oct 21. Nat Rev Microbiol. 2021. PMID: 33087881 Free PMC article. Review.
-
Acidic pH Mediates Changes in Antigenic and Oligomeric Conformation of Herpes Simplex Virus gB and Is a Determinant of Cell-Specific Entry.J Virol. 2018 Aug 16;92(17):e01034-18. doi: 10.1128/JVI.01034-18. Print 2018 Sep 1. J Virol. 2018. PMID: 29925660 Free PMC article.
References
-
- Cocchi F, Menotti L, Mirandola P, Lopez M, Campadelli-Fiume G. 1998. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J Virol 72:9992–10002. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
