

The mechanism of activation of IRAK1 and IRAK4 by interleukin-1 and Toll-like receptor agonists
- PMID: 28512203
- PMCID: PMC5460469
- DOI: 10.1042/BCJ20170097
The mechanism of activation of IRAK1 and IRAK4 by interleukin-1 and Toll-like receptor agonists
Abstract
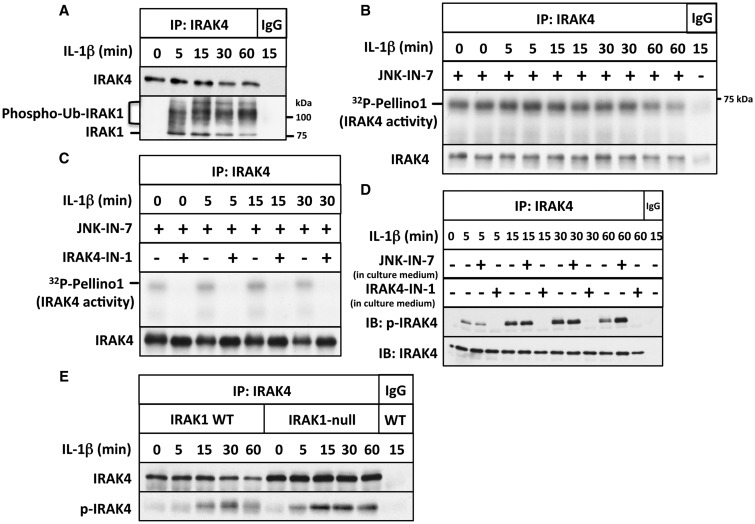
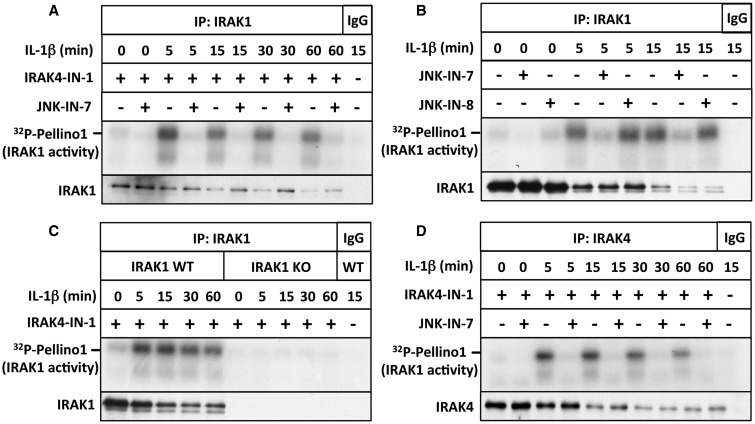
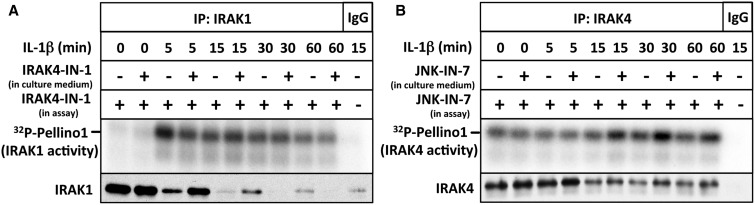
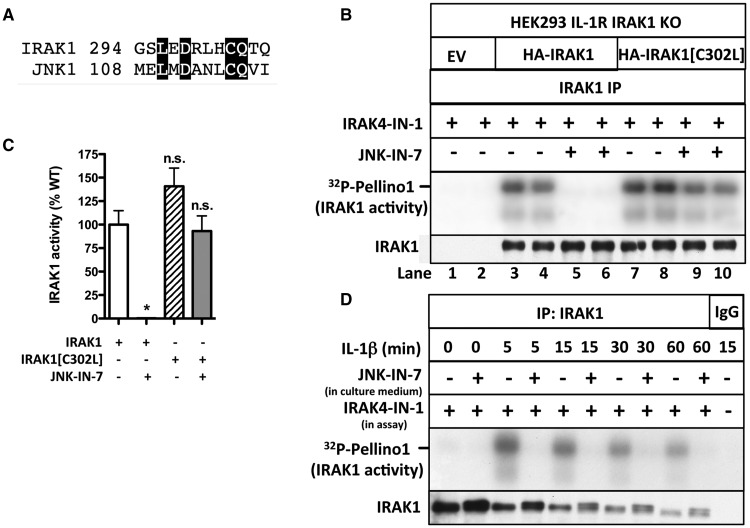
We have developed the first assays that measure the protein kinase activities of interleukin-1 receptor-associated kinase 1 (IRAK1) and IRAK4 reliably in human cell extracts, by employing Pellino1 as a substrate in conjunction with specific pharmacological inhibitors of IRAK1 and IRAK4. We exploited these assays to show that IRAK4 was constitutively active and that its intrinsic activity towards Pellino1 was not increased significantly by stimulation with interleukin-1 (IL-1) in IL-1R-expressing HEK293 cells, Pam3CSK4-stimulated human THP1 monocytes or primary human macrophages. Our results, in conjunction with those of other investigators, suggest that the IL-1-stimulated trans-autophosphorylation of IRAK4 is initiated by the myeloid differentiation primary response gene 88-induced dimerization of IRAK4 and is not caused by an increase in the intrinsic catalytic activity of IRAK4. In contrast with IRAK4, we found that IRAK1 was inactive in unstimulated cells and converted into an active protein kinase in response to IL-1 or Pam3CSK4 in human cells. Surprisingly, the IL-1-stimulated activation of IRAK1 was not affected by pharmacological inhibition of IRAK4 and not reversed by dephosphorylation and/or deubiquitylation, suggesting that IRAK1 catalytic activity is not triggered by a covalent modification but by an allosteric mechanism induced by its interaction with IRAK4.
Keywords: IRAK; MyD88; Pellino; Toll-like receptor; innate immunity.
© 2017 The Author(s).
Conflict of interest statement
The Authors declare that there are no competing interests associated with the manuscript.
Figures
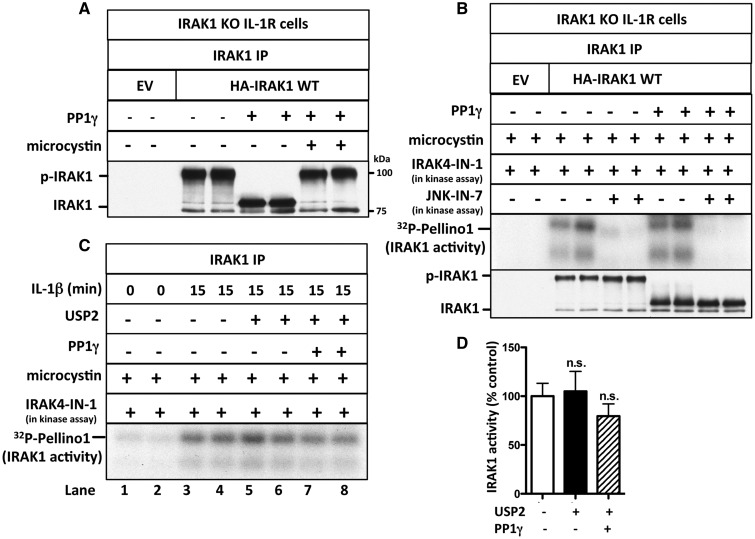
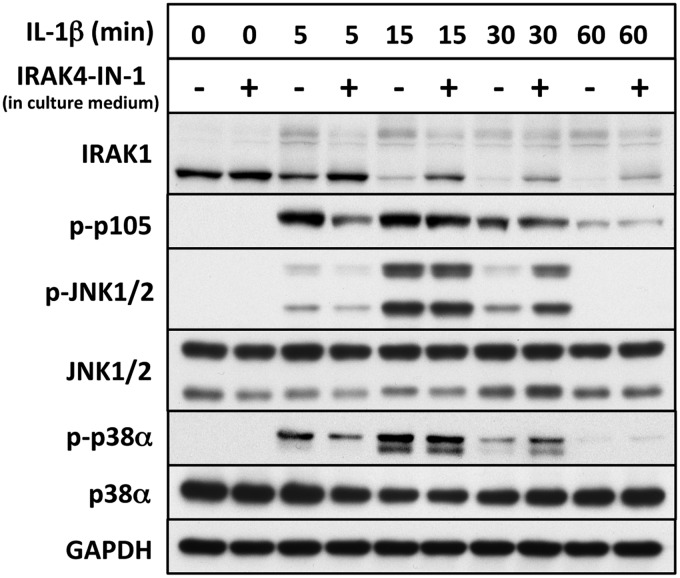
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous