

Engineering and Functional Characterization of Fusion Genes Identifies Novel Oncogenic Drivers of Cancer
- PMID: 28512244
- PMCID: PMC5568774
- DOI: 10.1158/0008-5472.CAN-16-2745
Engineering and Functional Characterization of Fusion Genes Identifies Novel Oncogenic Drivers of Cancer
Abstract
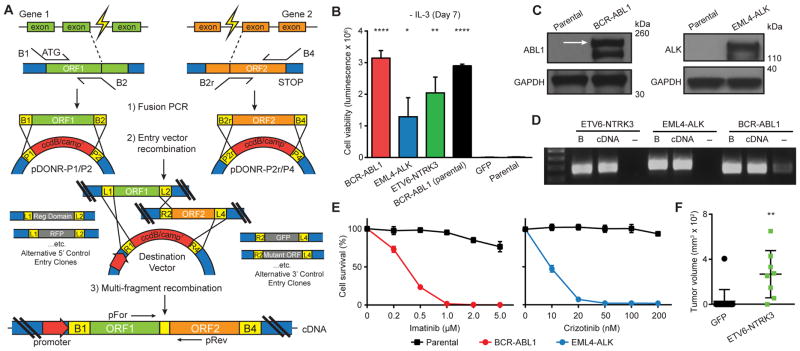
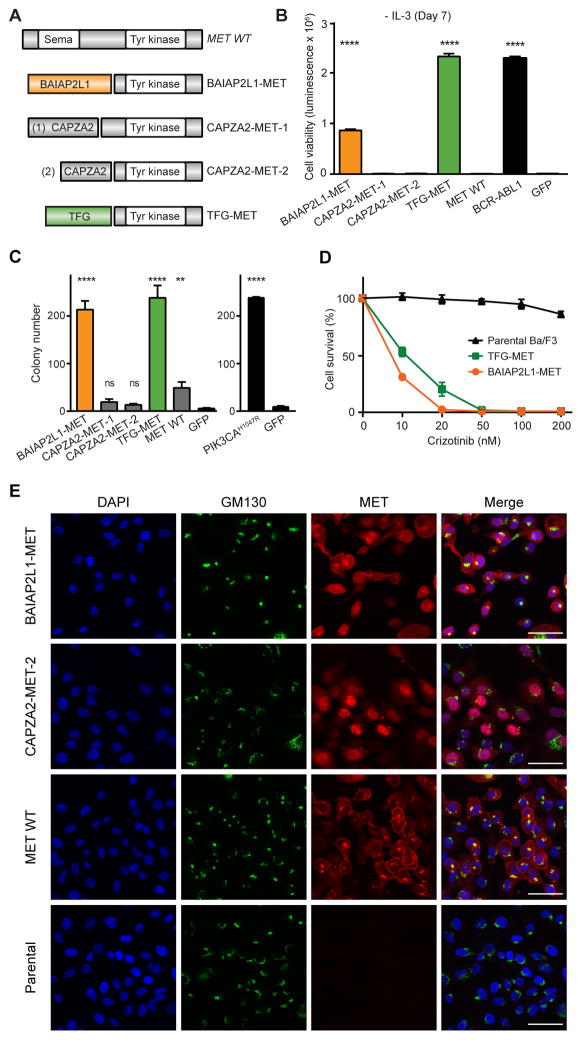
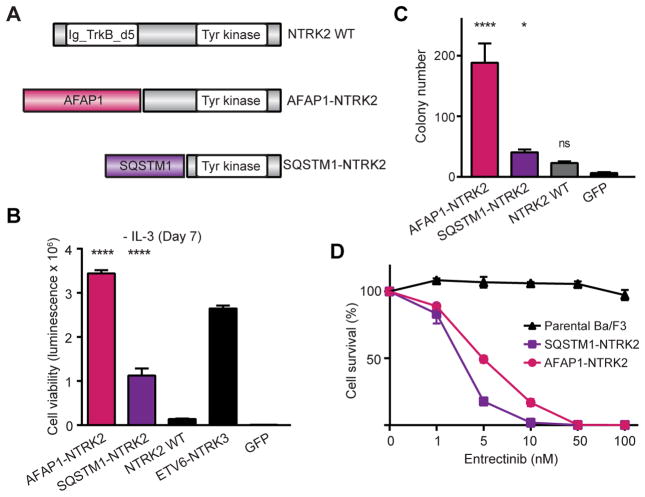
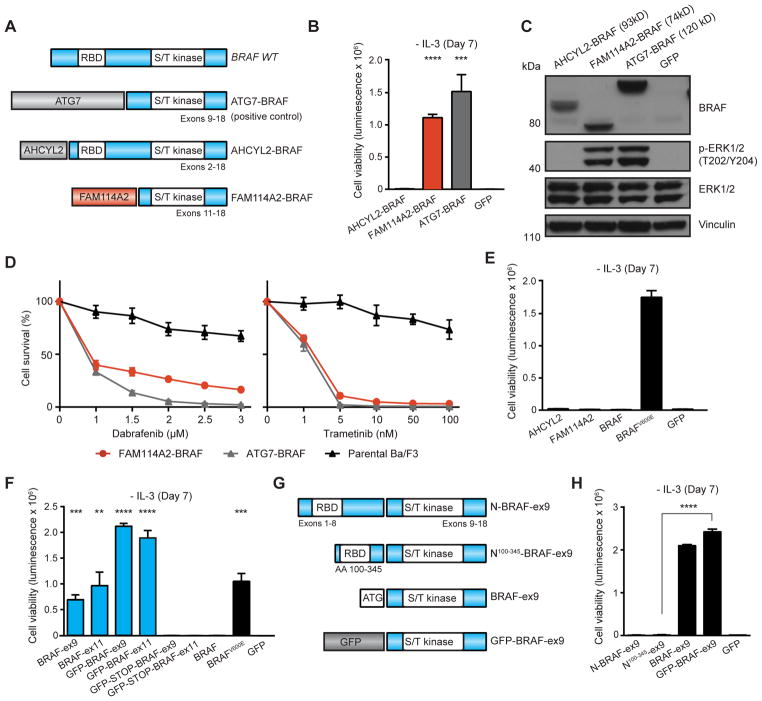
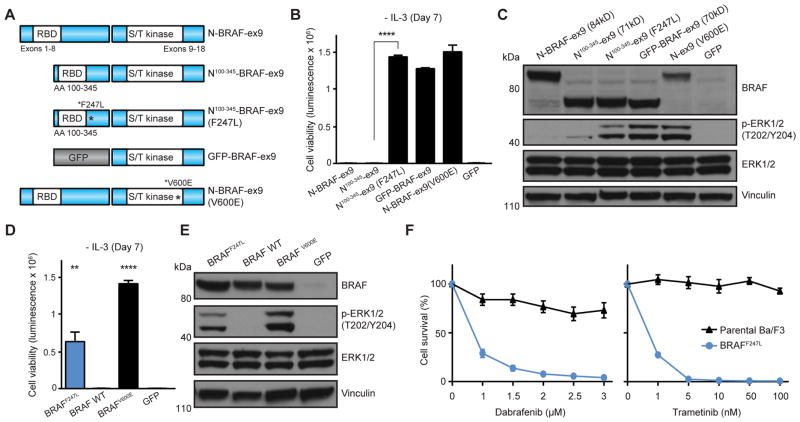
Oncogenic gene fusions drive many human cancers, but tools to more quickly unravel their functional contributions are needed. Here we describe methodology permitting fusion gene construction for functional evaluation. Using this strategy, we engineered the known fusion oncogenes, BCR-ABL1, EML4-ALK, and ETV6-NTRK3, as well as 20 previously uncharacterized fusion genes identified in The Cancer Genome Atlas datasets. In addition to confirming oncogenic activity of the known fusion oncogenes engineered by our construction strategy, we validated five novel fusion genes involving MET, NTRK2, and BRAF kinases that exhibited potent transforming activity and conferred sensitivity to FDA-approved kinase inhibitors. Our fusion construction strategy also enabled domain-function studies of BRAF fusion genes. Our results confirmed other reports that the transforming activity of BRAF fusions results from truncation-mediated loss of inhibitory domains within the N-terminus of the BRAF protein. BRAF mutations residing within this inhibitory region may provide a means for BRAF activation in cancer, therefore we leveraged the modular design of our fusion gene construction methodology to screen N-terminal domain mutations discovered in tumors that are wild-type at the BRAF mutation hotspot, V600. We identified an oncogenic mutation, F247L, whose expression robustly activated the MAPK pathway and sensitized cells to BRAF and MEK inhibitors. When applied broadly, these tools will facilitate rapid fusion gene construction for subsequent functional characterization and translation into personalized treatment strategies. Cancer Res; 77(13); 3502-12. ©2017 AACR.
©2017 American Association for Cancer Research.
Figures
References
-
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nature reviews Cancer. 2007;7:233–45. - PubMed
-
- Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362:2260–70. - PubMed
-
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. - PubMed
-
- Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–94. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
