

Targetable T-type Calcium Channels Drive Glioblastoma
- PMID: 28512247
- PMCID: PMC5505315
- DOI: 10.1158/0008-5472.CAN-16-2347
Targetable T-type Calcium Channels Drive Glioblastoma
Abstract
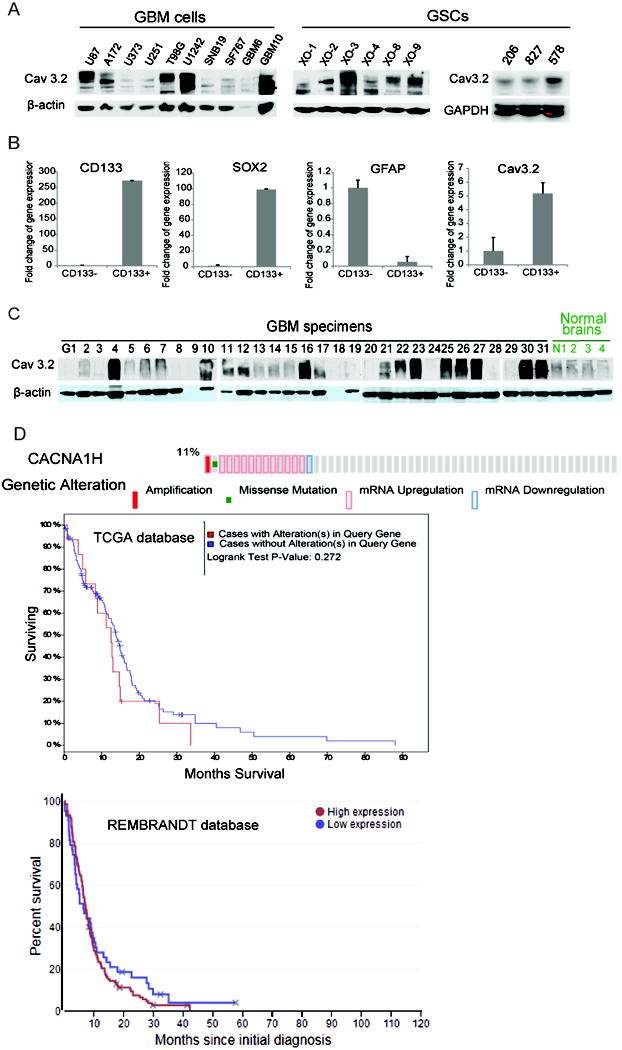
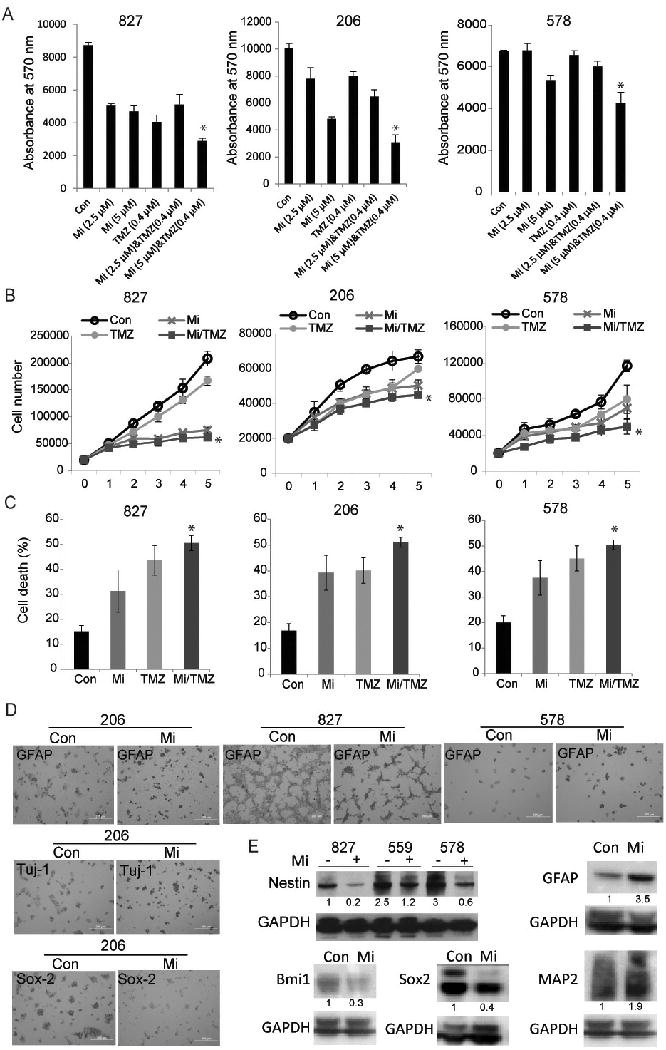
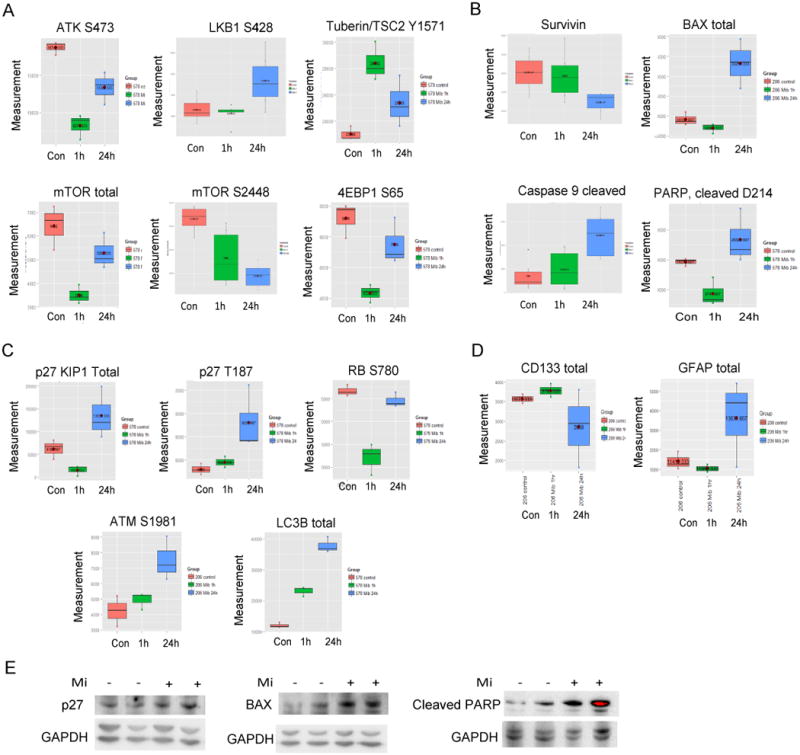
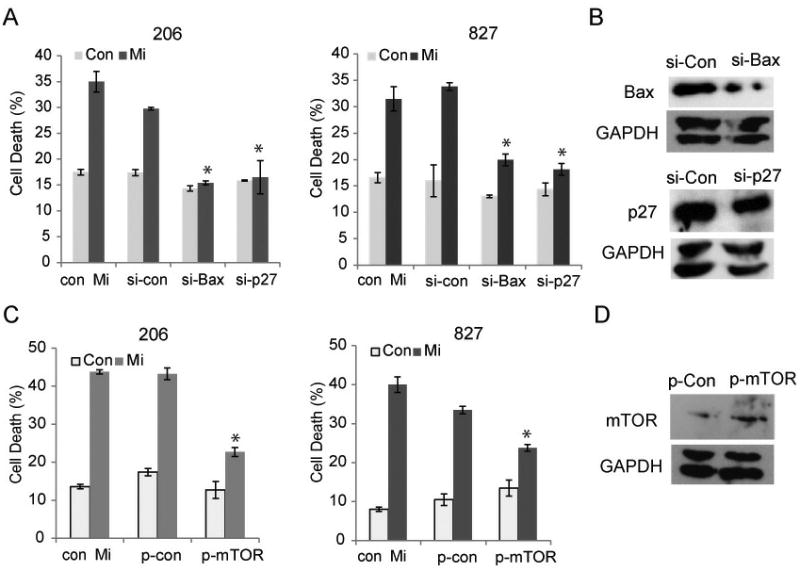
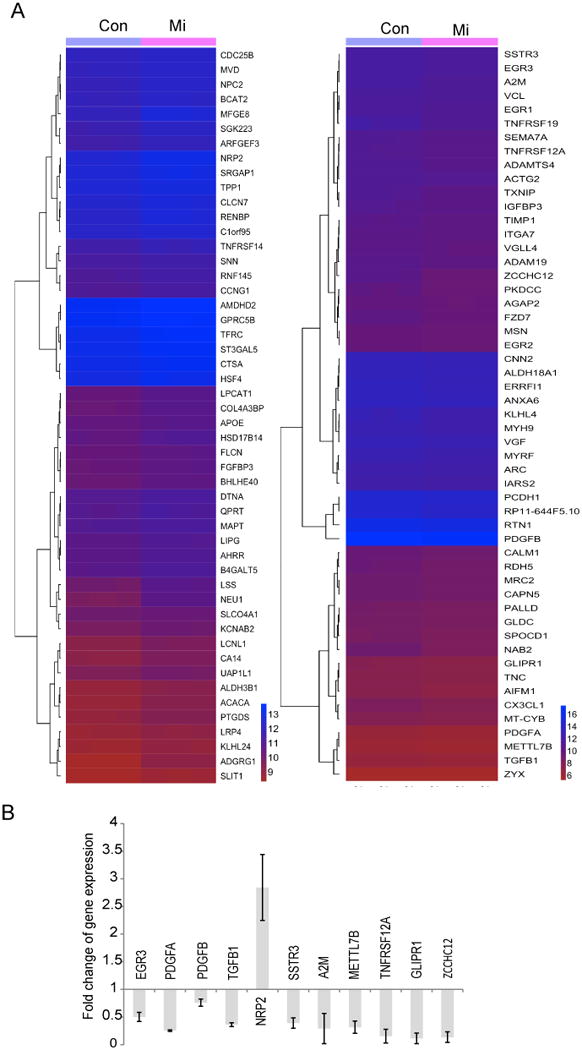
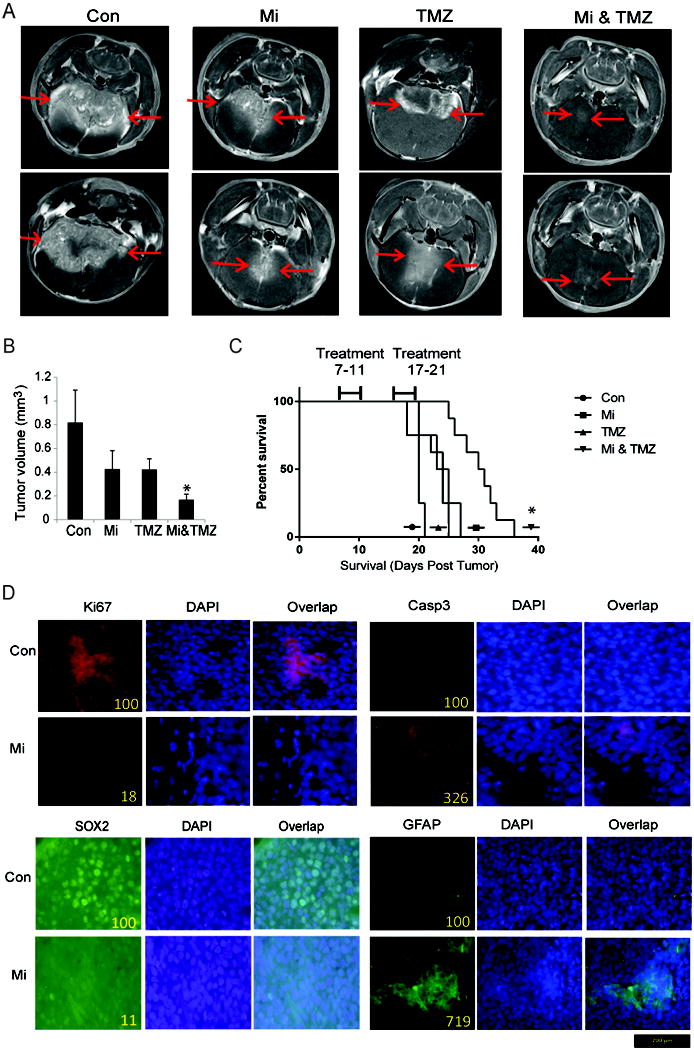
Glioblastoma (GBM) stem-like cells (GSC) promote tumor initiation, progression, and therapeutic resistance. Here, we show how GSCs can be targeted by the FDA-approved drug mibefradil, which inhibits the T-type calcium channel Cav3.2. This calcium channel was highly expressed in human GBM specimens and enriched in GSCs. Analyses of the The Cancer Genome Atlas and REMBRANDT databases confirmed upregulation of Cav3.2 in a subset of tumors and showed that overexpression associated with worse prognosis. Mibefradil treatment or RNAi-mediated attenuation of Cav3.2 was sufficient to inhibit the growth, survival, and stemness of GSCs and also sensitized them to temozolomide chemotherapy. Proteomic and transcriptomic analyses revealed that Cav3.2 inhibition altered cancer signaling pathways and gene transcription. Cav3.2 inhibition suppressed GSC growth in part by inhibiting prosurvival AKT/mTOR pathways and stimulating proapoptotic survivin and BAX pathways. Furthermore, Cav3.2 inhibition decreased expression of oncogenes (PDGFA, PDGFB, and TGFB1) and increased expression of tumor suppressor genes (TNFRSF14 and HSD17B14). Oral administration of mibefradil inhibited growth of GSC-derived GBM murine xenografts, prolonged host survival, and sensitized tumors to temozolomide treatment. Our results offer a comprehensive characterization of Cav3.2 in GBM tumors and GSCs and provide a preclinical proof of concept for repurposing mibefradil as a mechanism-based treatment strategy for GBM. Cancer Res; 77(13); 3479-90. ©2017 AACR.
©2017 American Association for Cancer Research.
Conflict of interest statement
Figures

References
-
- Wen PY, Reardon DA. Neuro-oncology in 2015: Progress in glioma diagnosis, classification and treatment. Nature reviews Neurology. 2016;12:69–70. - PubMed
-
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer research. 2004;64:7011–21. - PubMed
-
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. - PubMed
-
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
