

Protein Homeostasis in Amyotrophic Lateral Sclerosis: Therapeutic Opportunities?
- PMID: 28512398
- PMCID: PMC5411428
- DOI: 10.3389/fnmol.2017.00123
Protein Homeostasis in Amyotrophic Lateral Sclerosis: Therapeutic Opportunities?
Abstract
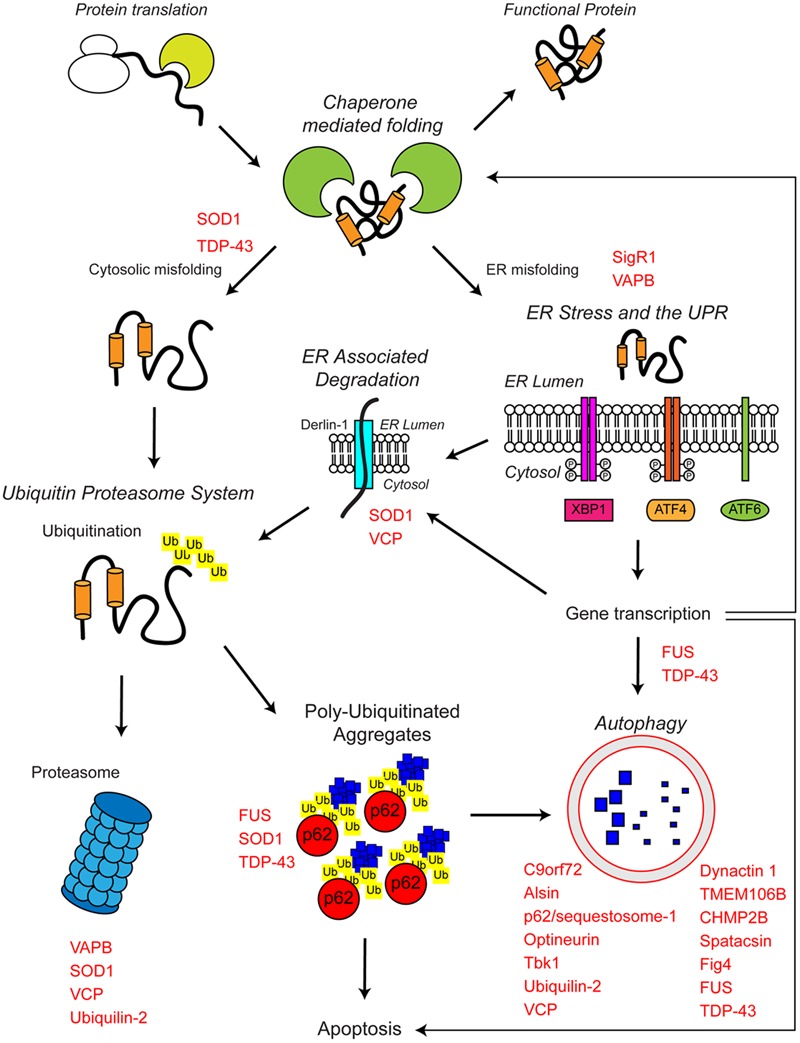
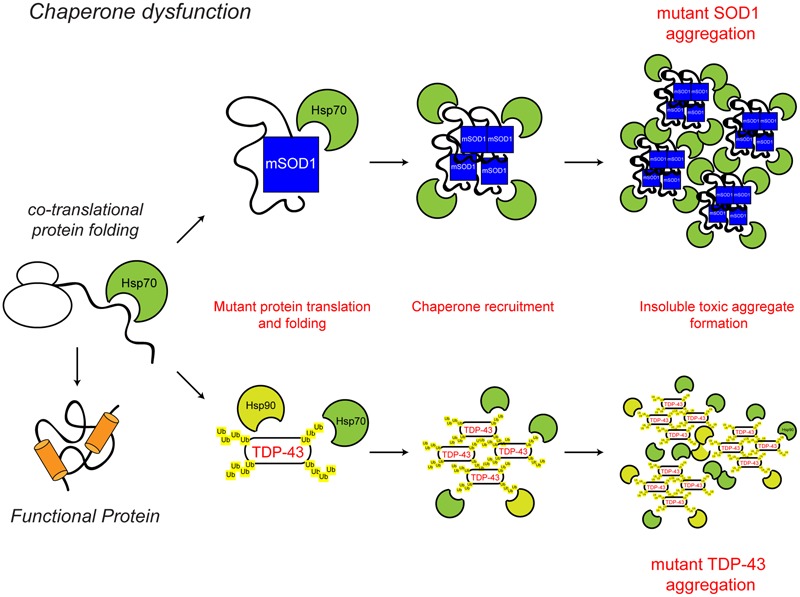
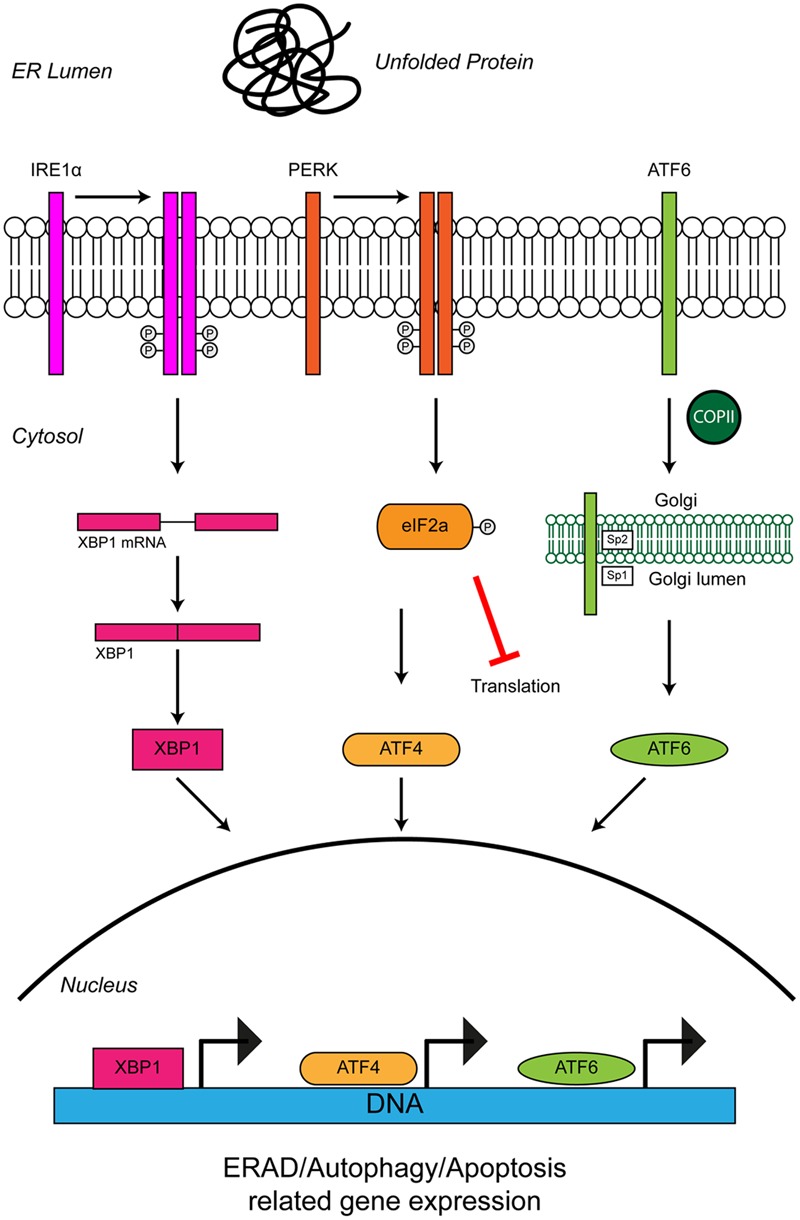
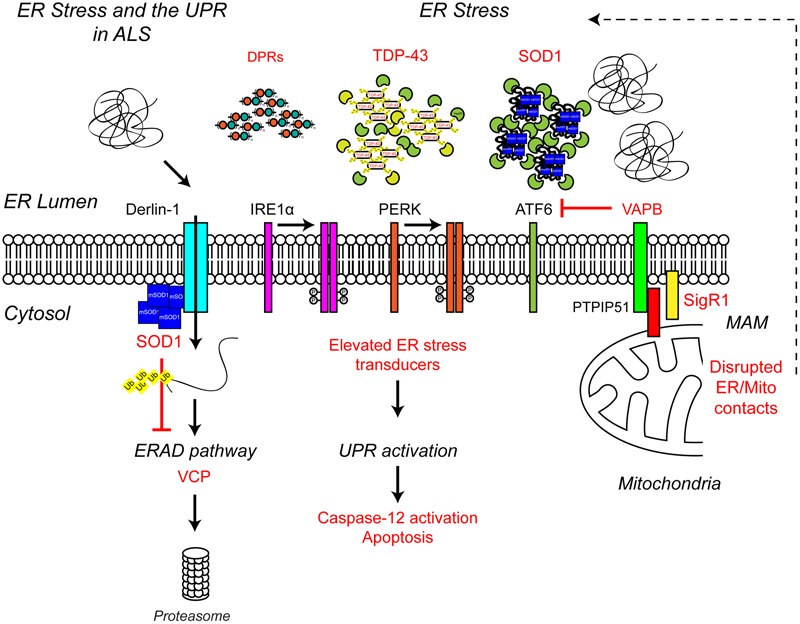
Protein homeostasis (proteostasis), the correct balance between production and degradation of proteins, is essential for the health and survival of cells. Proteostasis requires an intricate network of protein quality control pathways (the proteostasis network) that work to prevent protein aggregation and maintain proteome health throughout the lifespan of the cell. Collapse of proteostasis has been implicated in the etiology of a number of neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), the most common adult onset motor neuron disorder. Here, we review the evidence linking dysfunctional proteostasis to the etiology of ALS and discuss how ALS-associated insults affect the proteostasis network. Finally, we discuss the potential therapeutic benefit of proteostasis network modulation in ALS.
Keywords: amyotrophic lateral sclerosis (ALS); autophagy; chaperonins; motor neuron disease; protein aggregation; protein homeostasis; proteostasis; unfolded protein response (UPR).
Figures
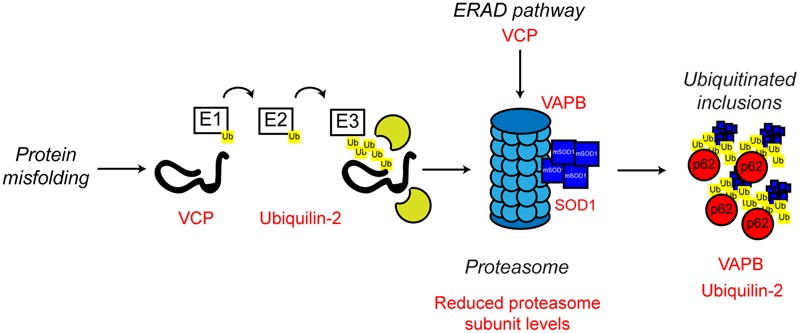
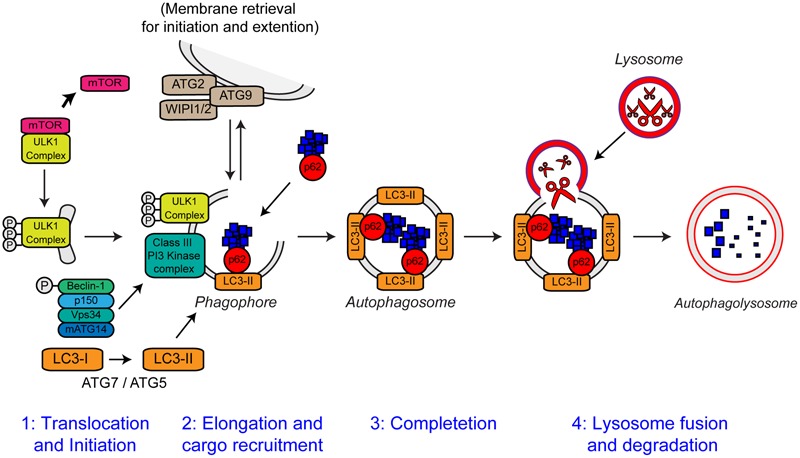
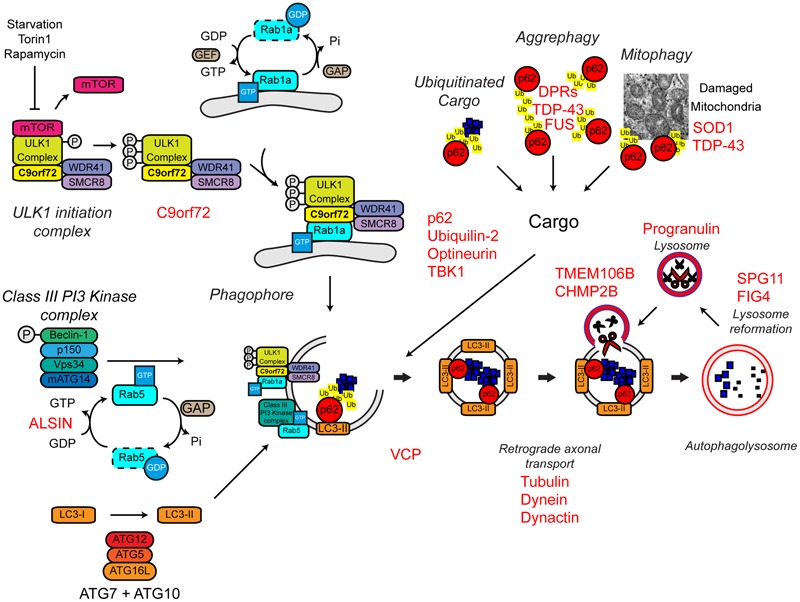
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous