

Architecture of the human interactome defines protein communities and disease networks
- PMID: 28514442
- PMCID: PMC5531611
- DOI: 10.1038/nature22366
Architecture of the human interactome defines protein communities and disease networks
Abstract
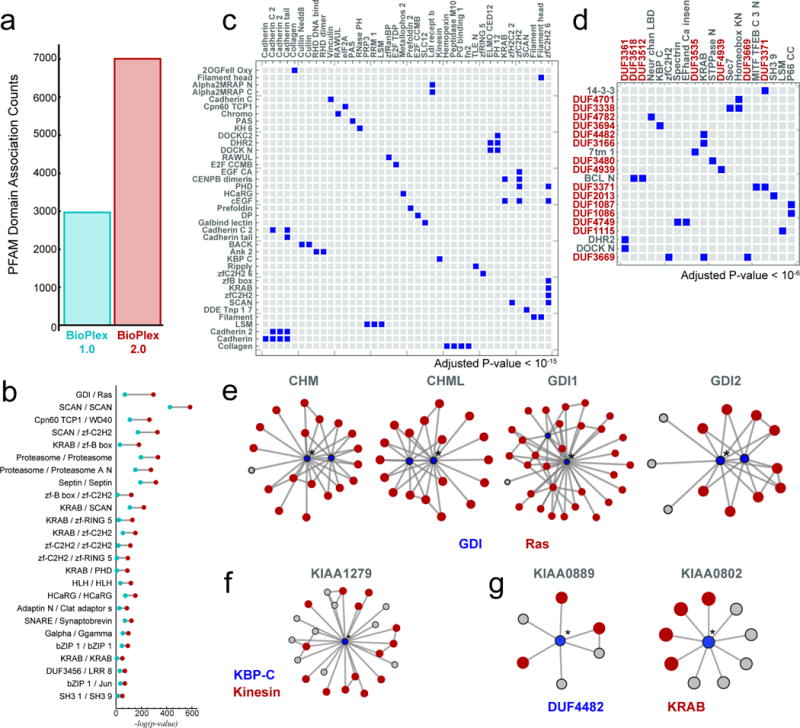
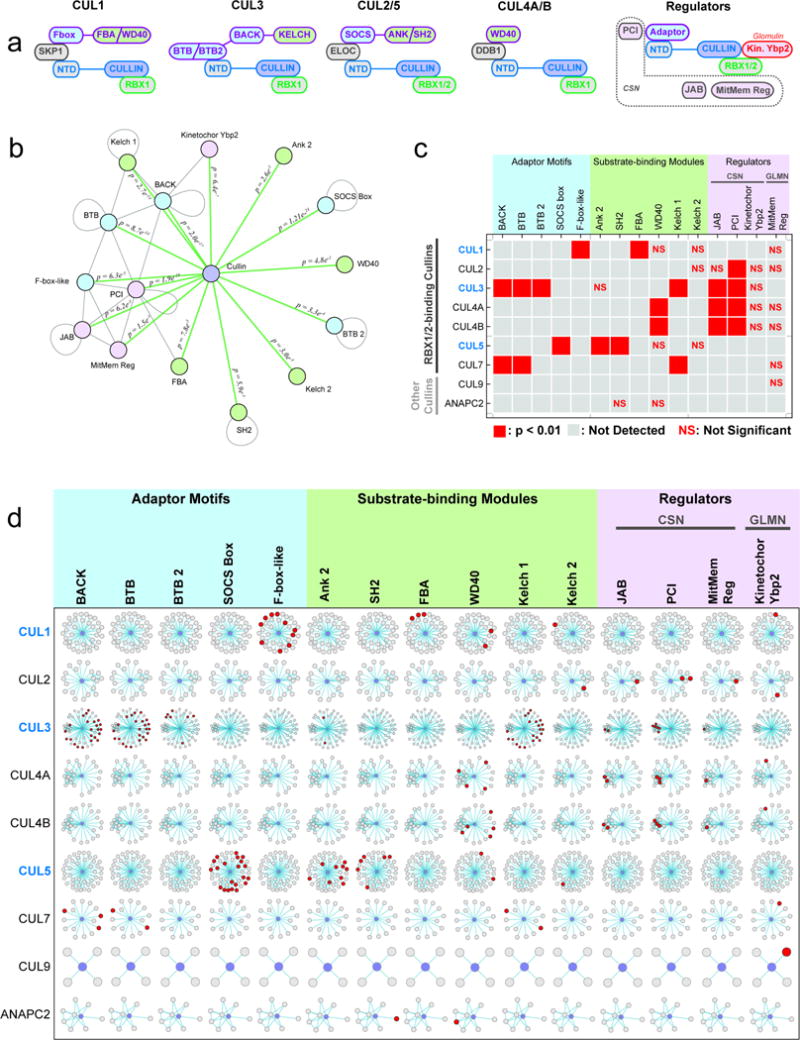
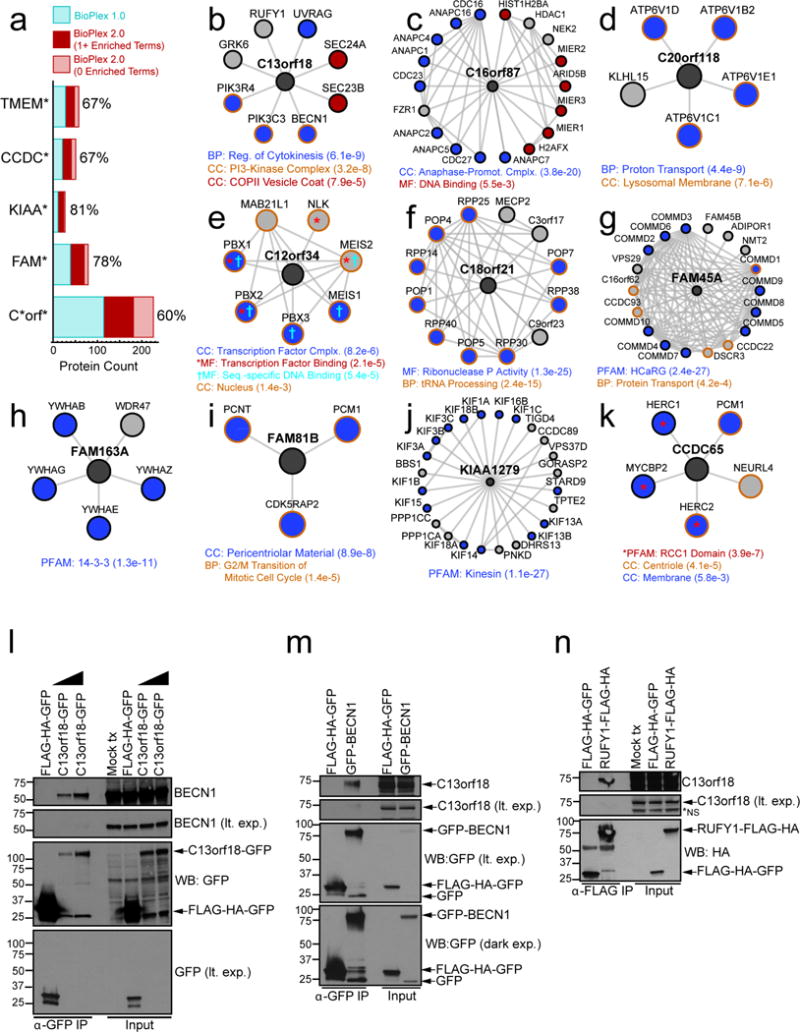
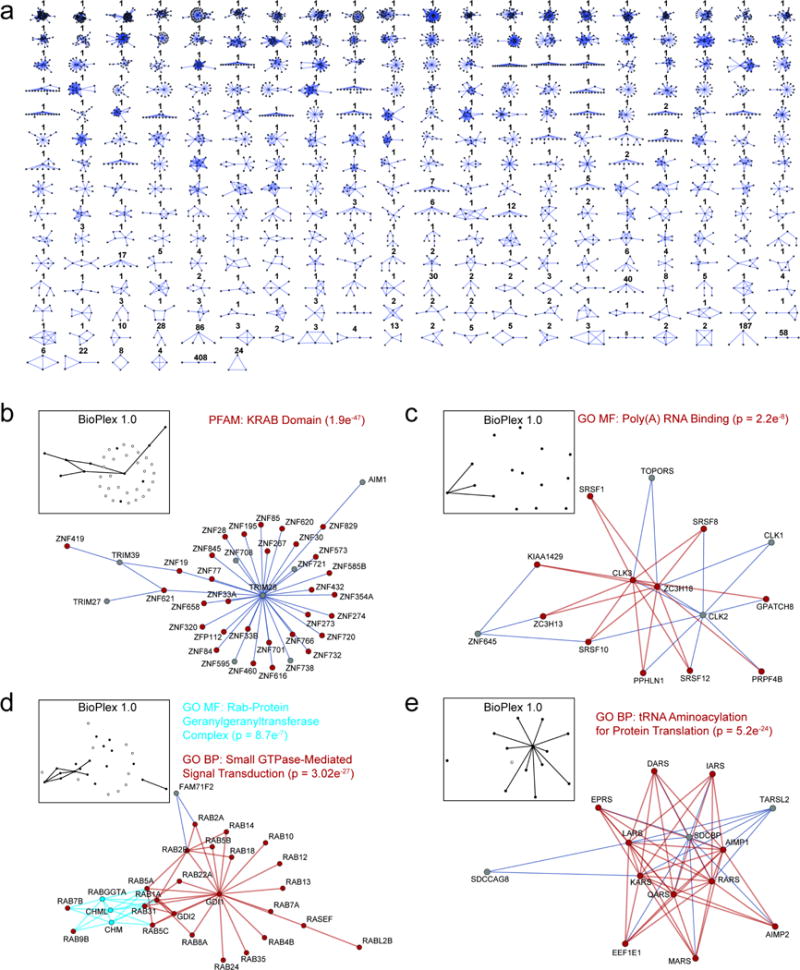
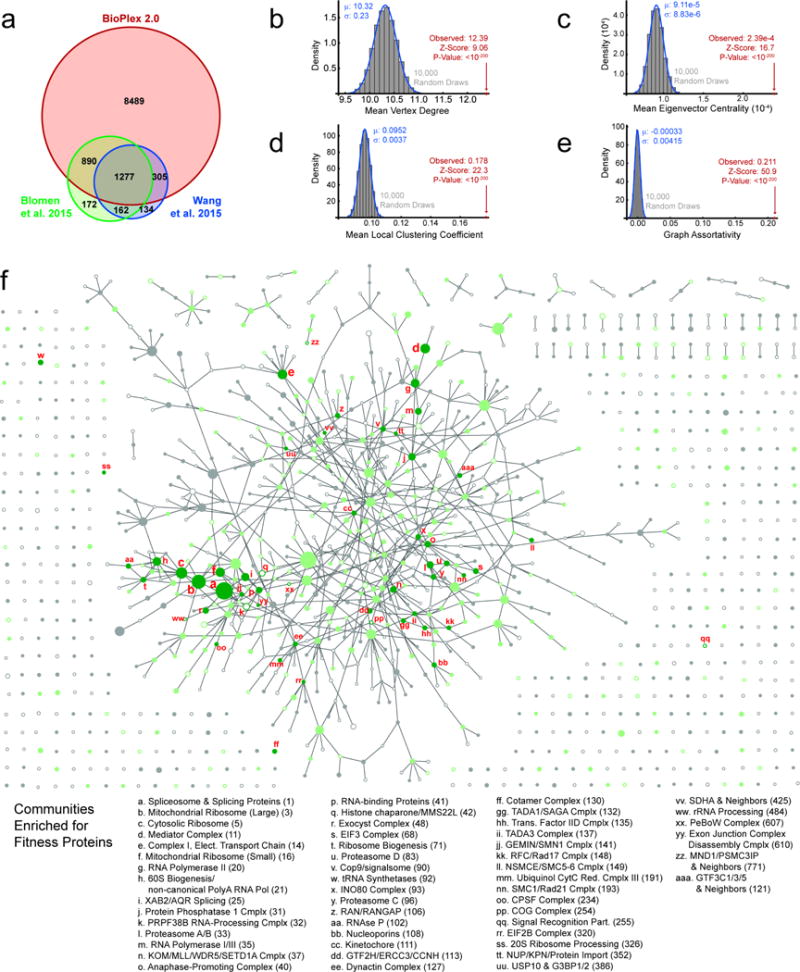
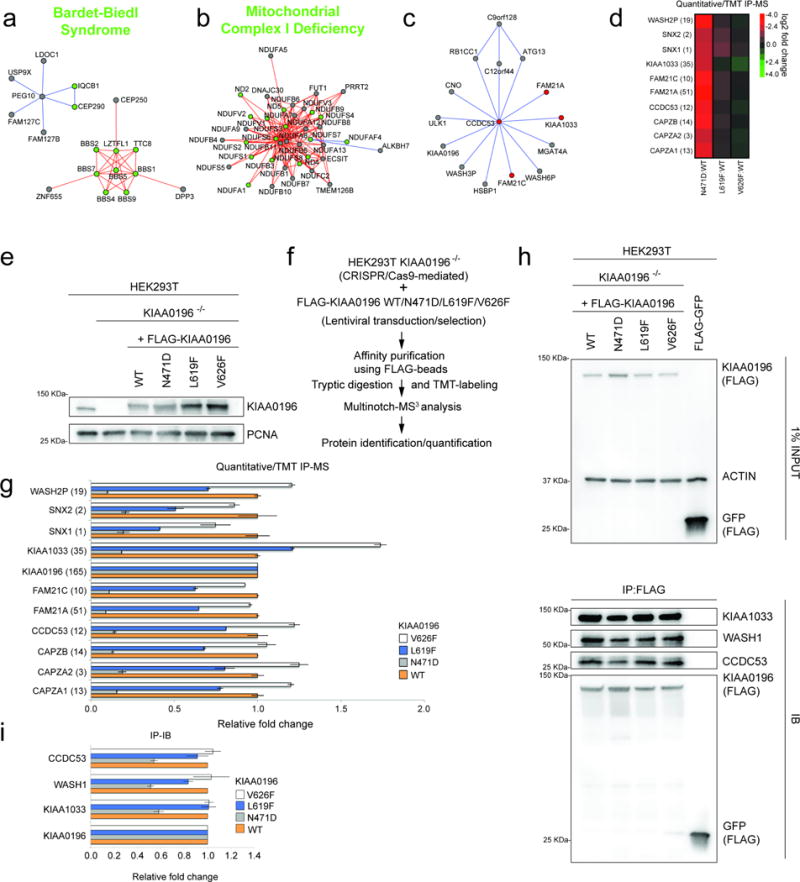
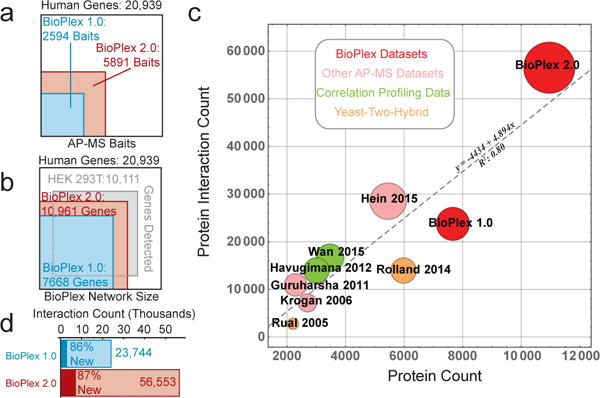
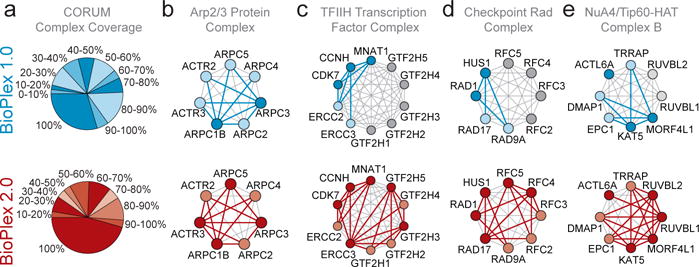
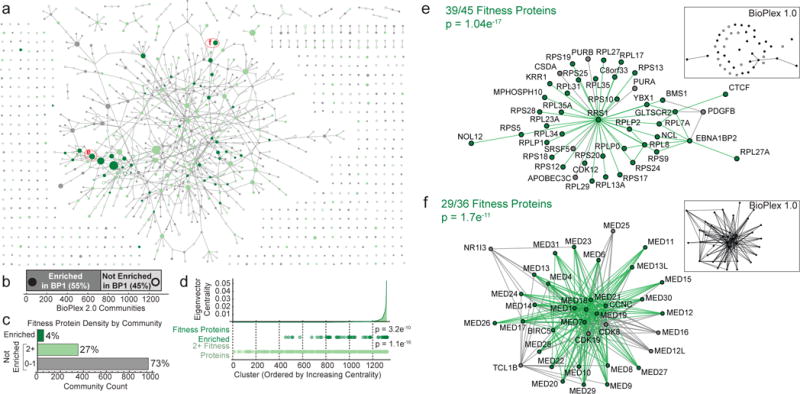
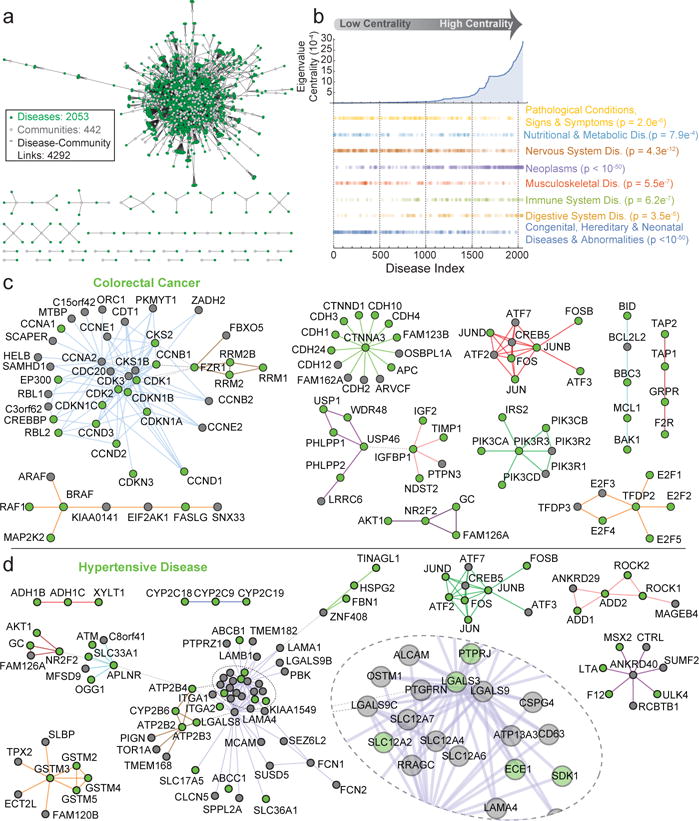
The physiology of a cell can be viewed as the product of thousands of proteins acting in concert to shape the cellular response. Coordination is achieved in part through networks of protein-protein interactions that assemble functionally related proteins into complexes, organelles, and signal transduction pathways. Understanding the architecture of the human proteome has the potential to inform cellular, structural, and evolutionary mechanisms and is critical to elucidating how genome variation contributes to disease. Here we present BioPlex 2.0 (Biophysical Interactions of ORFeome-derived complexes), which uses robust affinity purification-mass spectrometry methodology to elucidate protein interaction networks and co-complexes nucleated by more than 25% of protein-coding genes from the human genome, and constitutes, to our knowledge, the largest such network so far. With more than 56,000 candidate interactions, BioPlex 2.0 contains more than 29,000 previously unknown co-associations and provides functional insights into hundreds of poorly characterized proteins while enhancing network-based analyses of domain associations, subcellular localization, and co-complex formation. Unsupervised Markov clustering of interacting proteins identified more than 1,300 protein communities representing diverse cellular activities. Genes essential for cell fitness are enriched within 53 communities representing central cellular functions. Moreover, we identified 442 communities associated with more than 2,000 disease annotations, placing numerous candidate disease genes into a cellular framework. BioPlex 2.0 exceeds previous experimentally derived interaction networks in depth and breadth, and will be a valuable resource for exploring the biology of incompletely characterized proteins and for elucidating larger-scale patterns of proteome organization.
Conflict of interest statement
Yes there is potential Competing Interest.
Steven Gygi is a consultant for Biogen, Inc.
Figures
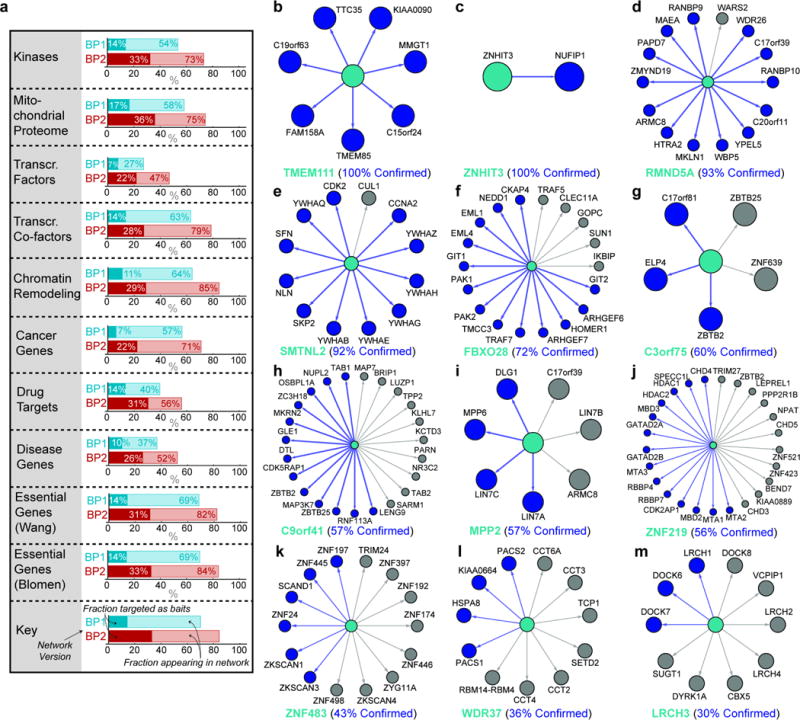
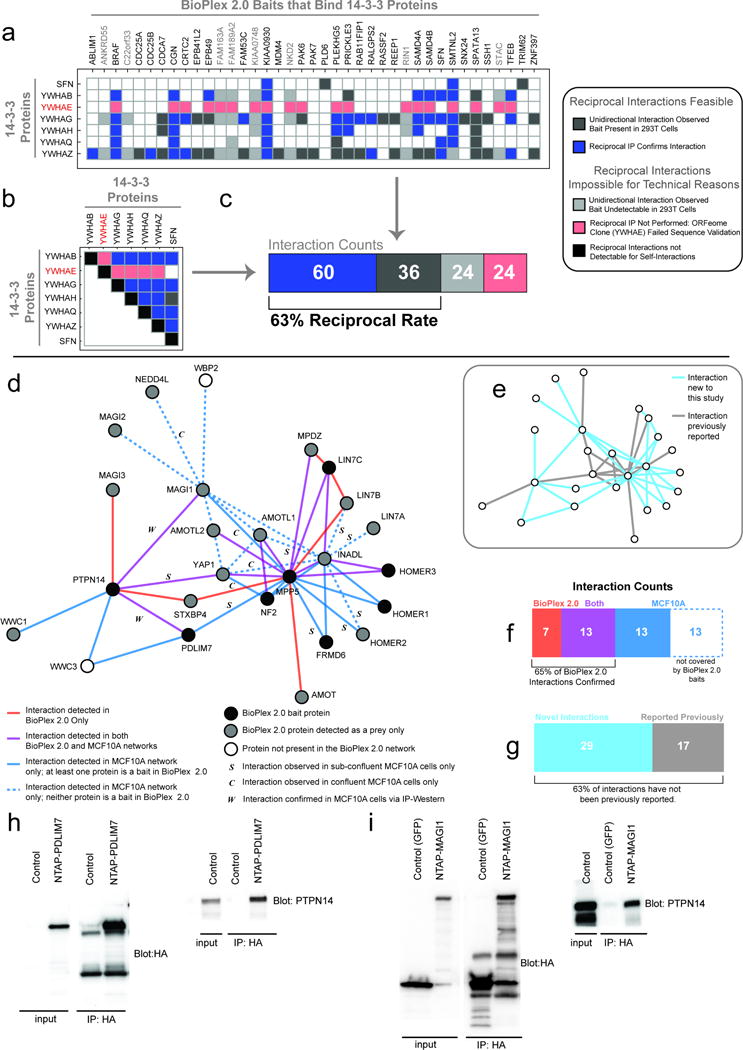
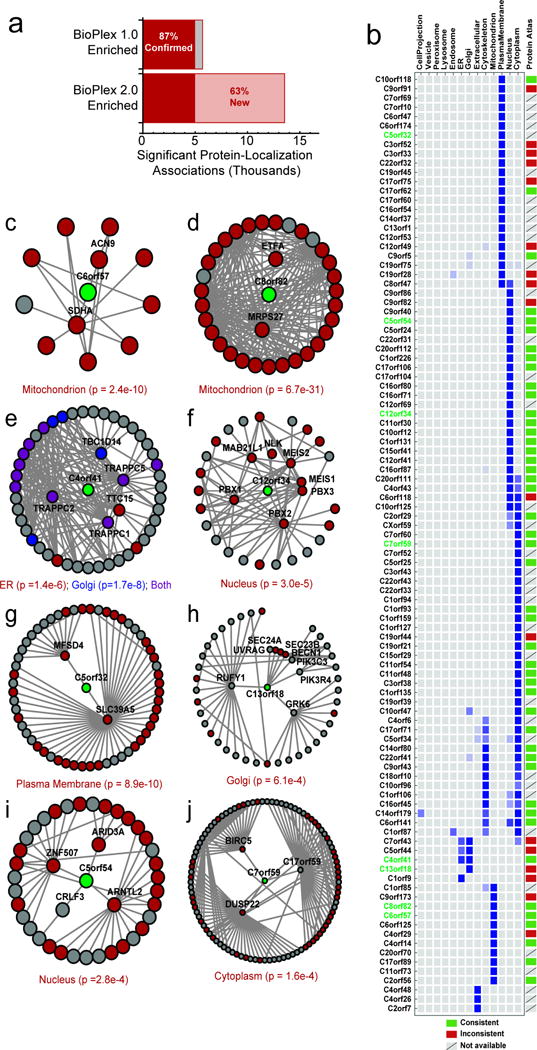
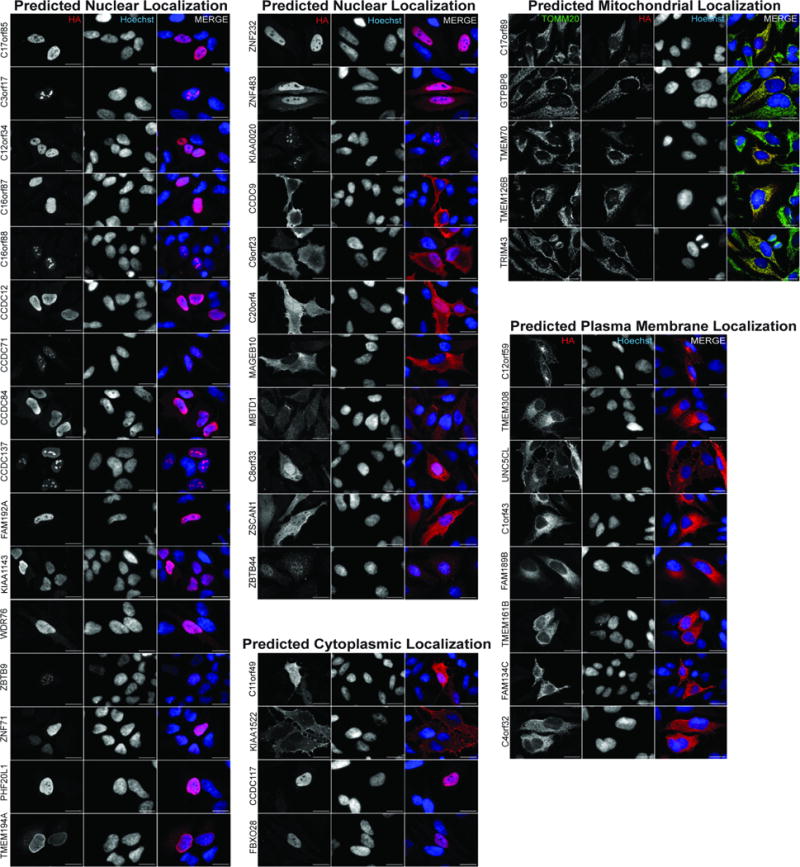
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
