

DNA-damage-induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair
- PMID: 28515316
- PMCID: PMC5491765
- DOI: 10.1074/jbc.M116.772475
DNA-damage-induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair
Abstract
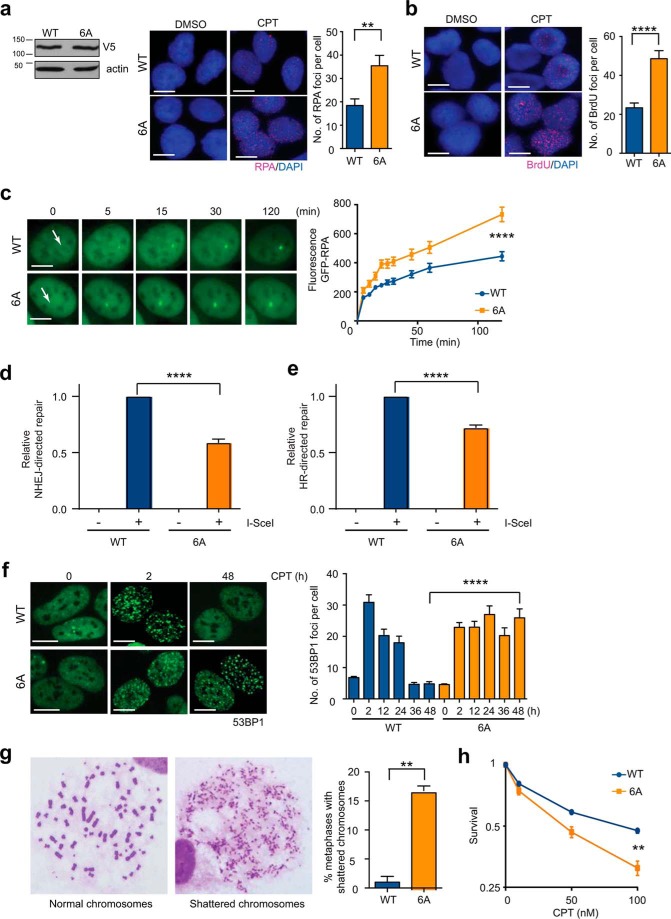
End resection of DNA double-strand breaks (DSBs) to generate 3'-single-stranded DNA facilitates DSB repair via error-free homologous recombination (HR) while stymieing repair by the error-prone non-homologous end joining (NHEJ) pathway. Activation of DNA end resection involves phosphorylation of the 5' to 3' exonuclease EXO1 by the phosphoinositide 3-kinase-like kinases ATM (ataxia telangiectasia-mutated) and ATR (ATM and Rad3-related) and by the cyclin-dependent kinases 1 and 2. After activation, EXO1 must also be restrained to prevent over-resection that is known to hamper optimal HR and trigger global genomic instability. However, mechanisms by which EXO1 is restrained are still unclear. Here, we report that EXO1 is rapidly degraded by the ubiquitin-proteasome system soon after DSB induction in human cells. ATR inhibition attenuated DNA-damage-induced EXO1 degradation, indicating that ATR-mediated phosphorylation of EXO1 targets it for degradation. In accord with these results, EXO1 became resistant to degradation when its SQ motifs required for ATR-mediated phosphorylation were mutated. We show that upon the induction of DNA damage, EXO1 is ubiquitinated by a member of the Skp1-Cullin1-F-box (SCF) family of ubiquitin ligases in a phosphorylation-dependent manner. Importantly, expression of degradation-resistant EXO1 resulted in hyper-resection, which attenuated both NHEJ and HR and severely compromised DSB repair resulting in chromosomal instability. These findings indicate that the coupling of EXO1 activation with its eventual degradation is a timing mechanism that limits the extent of DNA end resection for accurate DNA repair.
Keywords: ATR; DNA double-strand break; DNA end resection; DNA repair; DNA-damage response; EXO1; Genomic stability; chromosomes; homologous recombination.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflict of interest with the contents of this article
Figures




Similar articles
-
DNA end resection is needed for the repair of complex lesions in G1-phase human cells.Cell Cycle. 2014;13(16):2509-16. doi: 10.4161/15384101.2015.941743. Cell Cycle. 2014. PMID: 25486192 Free PMC article.
-
Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks.Nucleic Acids Res. 2010 Apr;38(6):1821-31. doi: 10.1093/nar/gkp1164. Epub 2009 Dec 17. Nucleic Acids Res. 2010. PMID: 20019063 Free PMC article.
-
RBX1 prompts degradation of EXO1 to limit the homologous recombination pathway of DNA double-strand break repair in G1 phase.Cell Death Differ. 2020 Apr;27(4):1383-1397. doi: 10.1038/s41418-019-0424-4. Epub 2019 Sep 27. Cell Death Differ. 2020. PMID: 31562368 Free PMC article.
-
Role of deubiquitinating enzymes in DNA double-strand break repair.J Zhejiang Univ Sci B. 2021 Jan 15;22(1):63-72. doi: 10.1631/jzus.B2000309. J Zhejiang Univ Sci B. 2021. PMID: 33448188 Free PMC article. Review.
-
An insight into understanding the coupling between homologous recombination mediated DNA repair and chromatin remodeling mechanisms in plant genome: an update.Cell Cycle. 2021 Sep;20(18):1760-1784. doi: 10.1080/15384101.2021.1966584. Epub 2021 Aug 26. Cell Cycle. 2021. PMID: 34437813 Free PMC article. Review.
Cited by
-
A game of substrates: replication fork remodeling and its roles in genome stability and chemo-resistance.Cell Stress. 2017 Dec;1(3):115-133. doi: 10.15698/cst2017.12.114. Epub 2017 Dec 5. Cell Stress. 2017. PMID: 29355244 Free PMC article.
-
SMARCAL1 ubiquitylation controls its association with RPA-coated ssDNA and promotes replication fork stability.PLoS Biol. 2024 Mar 19;22(3):e3002552. doi: 10.1371/journal.pbio.3002552. eCollection 2024 Mar. PLoS Biol. 2024. PMID: 38502677 Free PMC article.
-
A single Ho-induced double-strand break at the MAT locus is lethal in Candida glabrata.PLoS Genet. 2020 Oct 15;16(10):e1008627. doi: 10.1371/journal.pgen.1008627. eCollection 2020 Oct. PLoS Genet. 2020. PMID: 33057400 Free PMC article.
-
Comparison of the different mechanisms of cytotoxicity induced by checkpoint kinase I inhibitors when used as single agents or in combination with DNA damage.Oncogene. 2020 Feb;39(7):1389-1401. doi: 10.1038/s41388-019-1079-9. Epub 2019 Oct 28. Oncogene. 2020. PMID: 31659257 Free PMC article. Review.
-
RPA Phosphorylation Inhibits DNA Resection.Mol Cell. 2019 Jul 11;75(1):145-153.e5. doi: 10.1016/j.molcel.2019.05.005. Epub 2019 May 29. Mol Cell. 2019. PMID: 31153714 Free PMC article.
References
-
- Burma S., Chen B. P., and Chen D. J. (2006) Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 5, 1042–1048 - PubMed
-
- Shrivastav M., De Haro L. P., and Nickoloff J. A. (2008) Regulation of DNA double-strand break repair pathway choice. Cell Res. 18, 134–147 - PubMed
-
- Chapman J. R., Taylor M. R., and Boulton S. J. (2012) Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47, 497–510 - PubMed
-
- Mladenov E., Magin S., Soni A., and Iliakis G. (2016) DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 37, 51–64 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
