

BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure
- PMID: 28515341
- PMCID: PMC5544253
- DOI: 10.1126/scitranslmed.aah5084
BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure
Erratum in
-
Erratum for the Research Article "BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure" by Q. Duan et al.Sci Transl Med. 2025 Feb 19;17(786):eadw3692. doi: 10.1126/scitranslmed.adw3692. Epub 2025 Feb 19. Sci Transl Med. 2025. PMID: 39970236 No abstract available.
Abstract
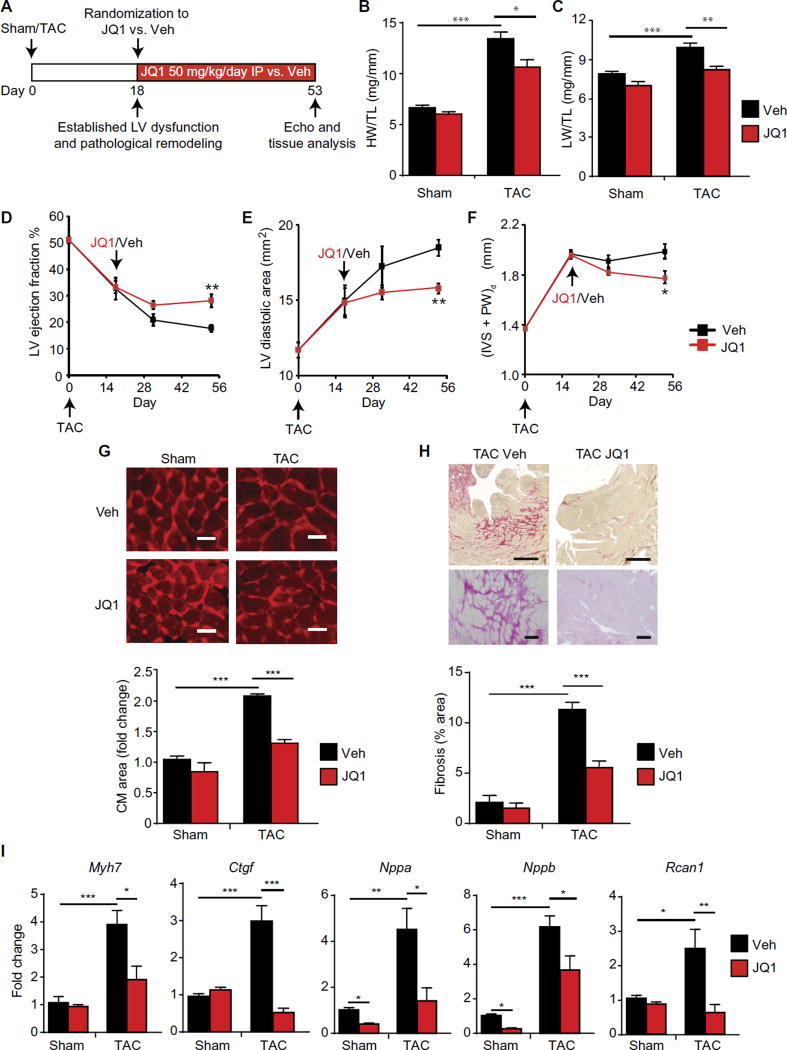
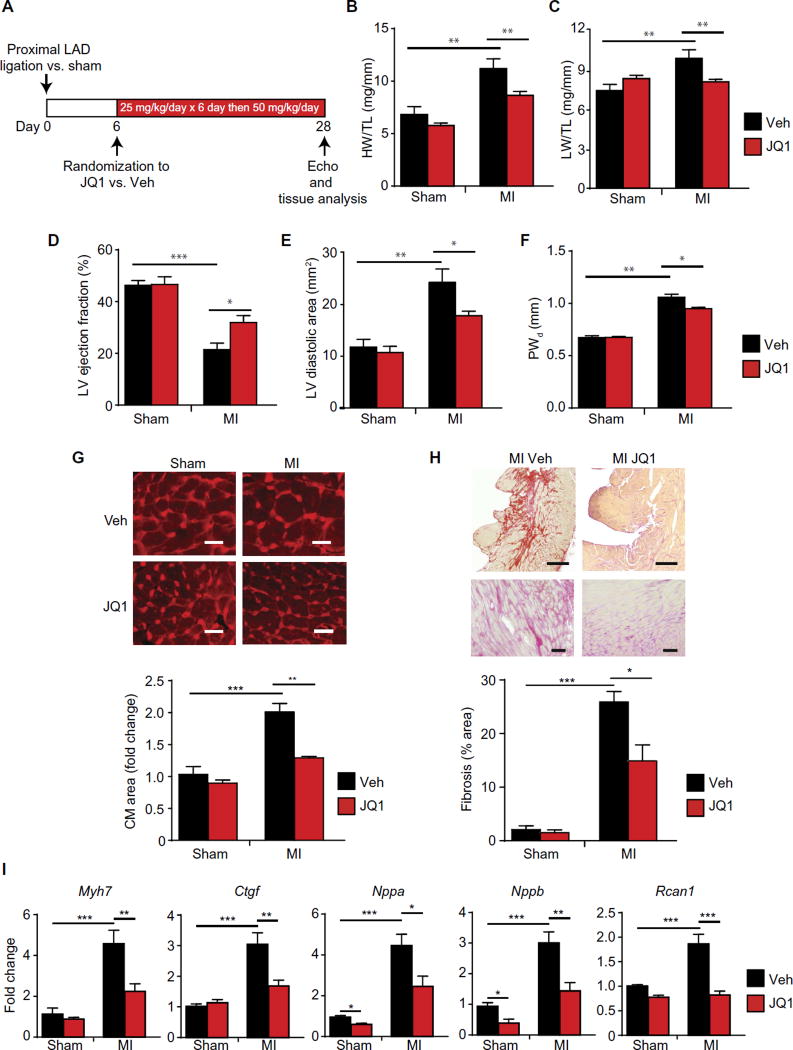
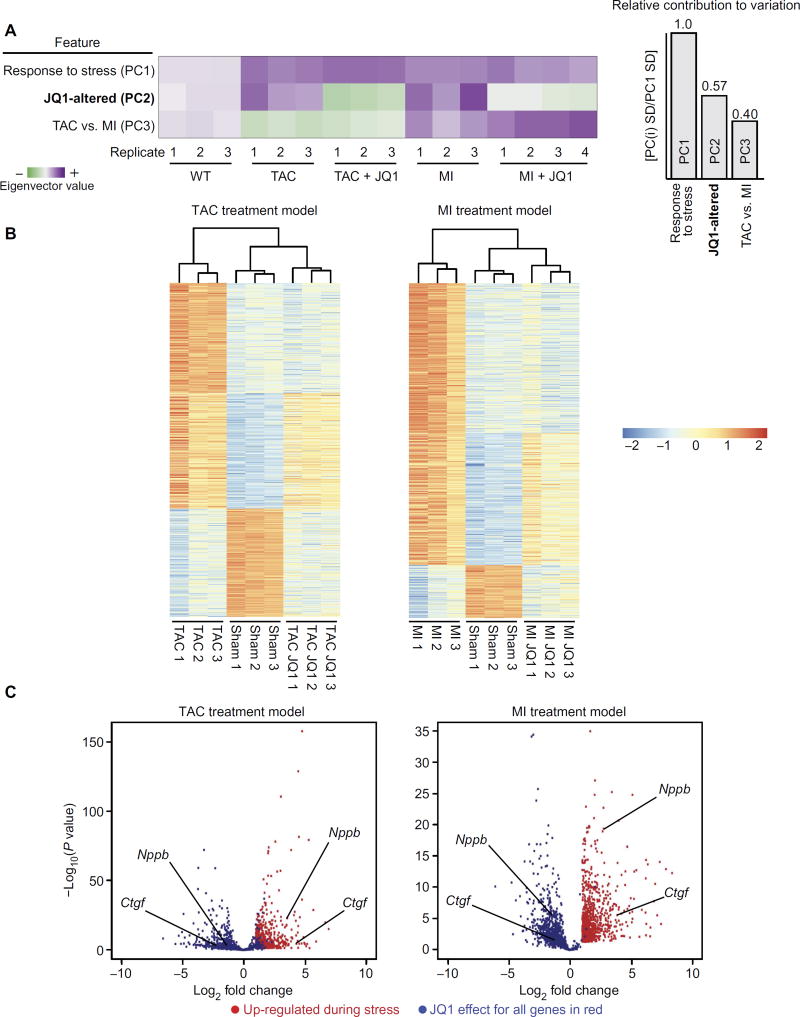
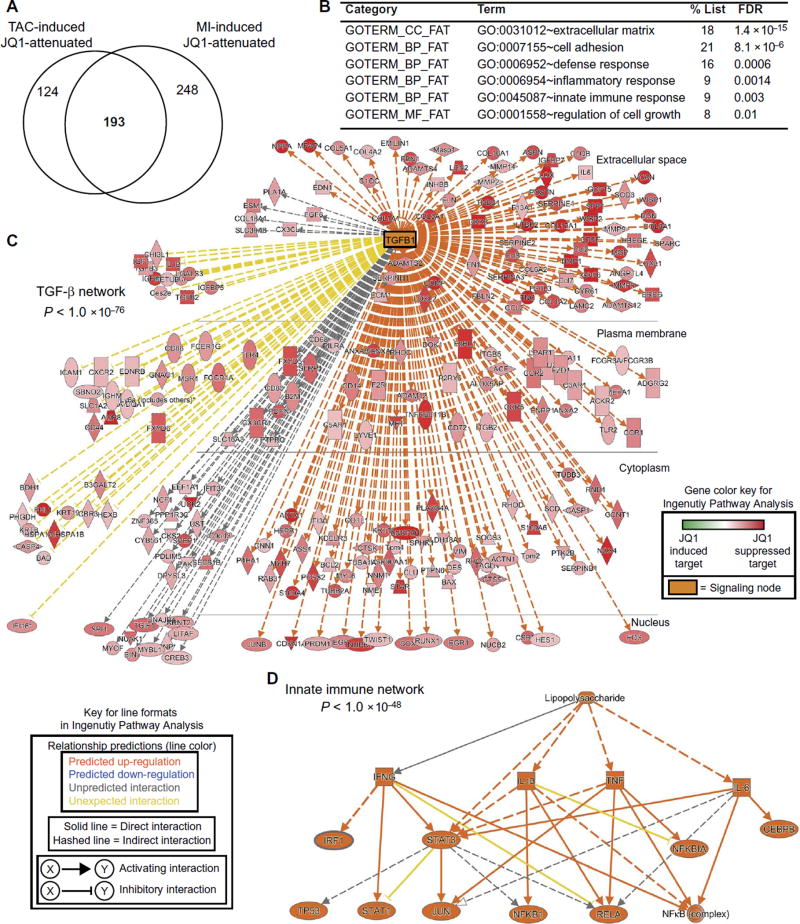
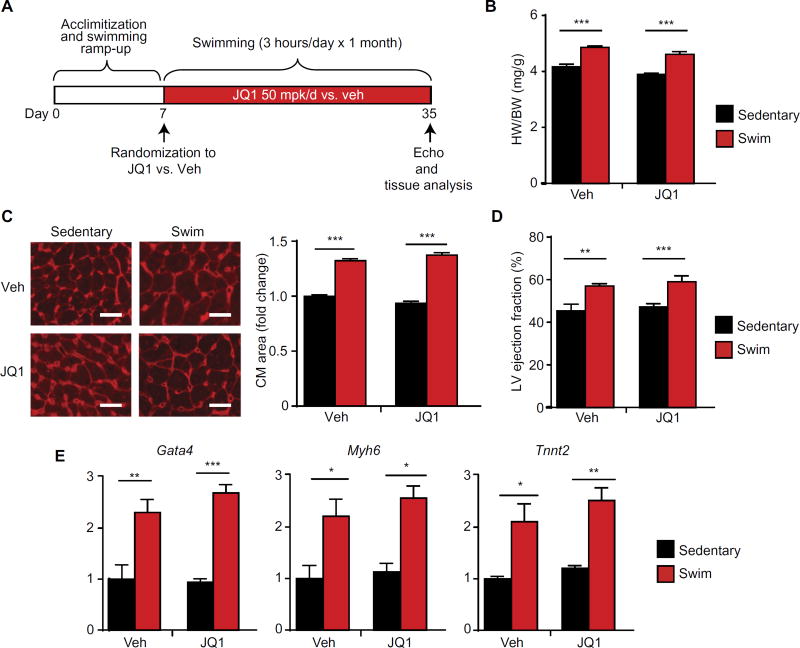
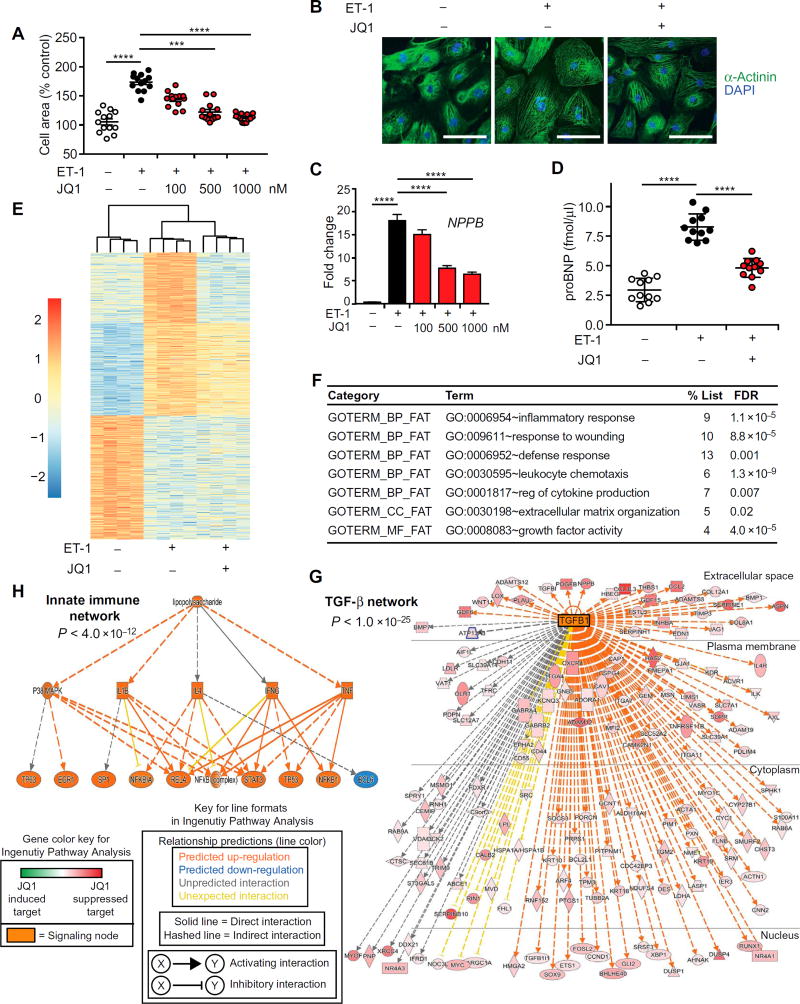
Despite current standard of care, the average 5-year mortality after an initial diagnosis of heart failure (HF) is about 40%, reflecting an urgent need for new therapeutic approaches. Previous studies demonstrated that the epigenetic reader protein bromodomain-containing protein 4 (BRD4), an emerging therapeutic target in cancer, functions as a critical coactivator of pathologic gene transactivation during cardiomyocyte hypertrophy. However, the therapeutic relevance of these findings to human disease remained unknown. We demonstrate that treatment with the BET bromodomain inhibitor JQ1 has therapeutic effects during severe, preestablished HF from prolonged pressure overload, as well as after a massive anterior myocardial infarction in mice. Furthermore, JQ1 potently blocks agonist-induced hypertrophy in human induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs). Integrated transcriptomic analyses across animal models and human iPSC-CMs reveal that BET inhibition preferentially blocks transactivation of a common pathologic gene regulatory program that is robustly enriched for NFκB and TGF-β signaling networks, typified by innate inflammatory and profibrotic myocardial genes. As predicted by these specific transcriptional mechanisms, we found that JQ1 does not suppress physiological cardiac hypertrophy in a mouse swimming model. These findings establish that pharmacologically targeting innate inflammatory and profibrotic myocardial signaling networks at the level of chromatin is effective in animal models and human cardiomyocytes, providing the critical rationale for further development of BET inhibitors and other epigenomic medicines for HF.
Copyright © 2017, American Association for the Advancement of Science.
Conflict of interest statement
Figures
Comment in
-
Cardiovascular disease: BET inhibitor attenuates heart failure.Nat Rev Drug Discov. 2017 Jun 29;16(7):453. doi: 10.1038/nrd.2017.125. Nat Rev Drug Discov. 2017. PMID: 28660902 No abstract available.
-
Letter to the editor: BET inhibitors might target innate inflammatory and profibrotic signaling networks in COPD.Am J Physiol Lung Cell Mol Physiol. 2017 Sep 1;313(3):L648-L649. doi: 10.1152/ajplung.00246.2017. Am J Physiol Lung Cell Mol Physiol. 2017. PMID: 28864534 No abstract available.
References
-
- Braunwald E. Research advances in heart failure: A compendium. Circ. Res. 2013;113:633–645. - PubMed
-
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Executive summary: Heart disease and stroke statistics—2012 update: A report from the American Heart Association. Circulation. 2012;125:188–197. - PubMed
-
- Hill JA, Olson EN. Cardiac plasticity. N. Engl. J. Med. 2008;358:1370–1380. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
