

Using high-resolution variant frequencies to empower clinical genome interpretation
- PMID: 28518168
- PMCID: PMC5563454
- DOI: 10.1038/gim.2017.26
Using high-resolution variant frequencies to empower clinical genome interpretation
Abstract
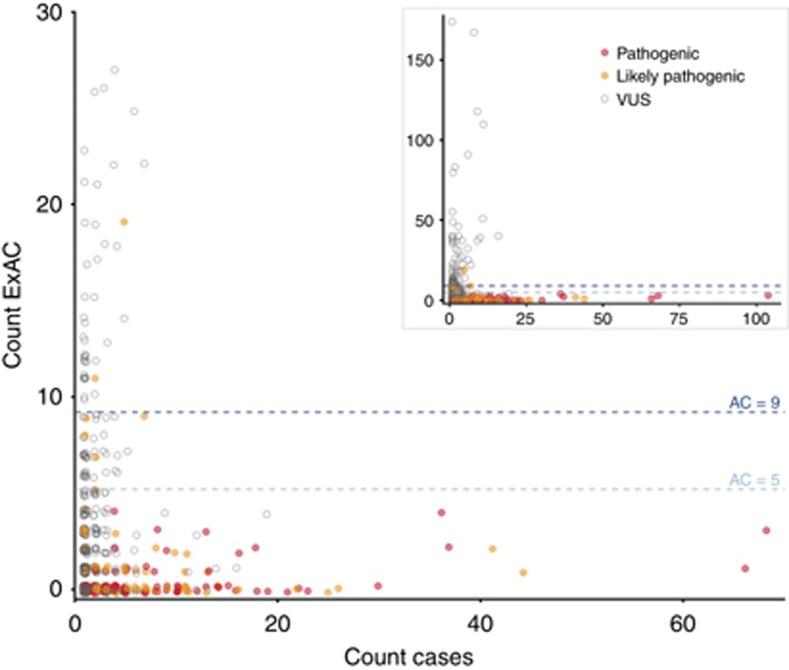
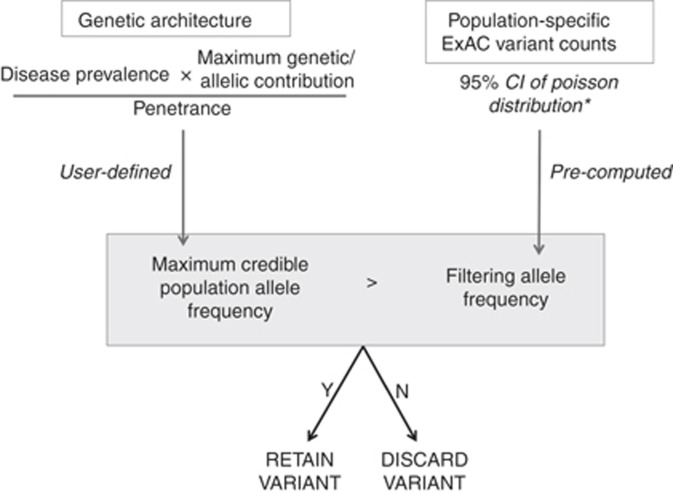
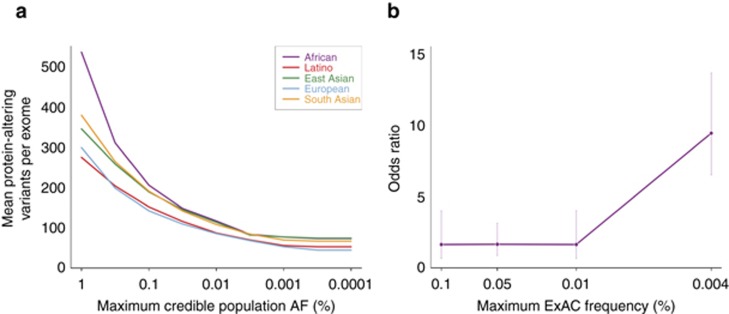
PurposeWhole-exome and whole-genome sequencing have transformed the discovery of genetic variants that cause human Mendelian disease, but discriminating pathogenic from benign variants remains a daunting challenge. Rarity is recognized as a necessary, although not sufficient, criterion for pathogenicity, but frequency cutoffs used in Mendelian analysis are often arbitrary and overly lenient. Recent very large reference datasets, such as the Exome Aggregation Consortium (ExAC), provide an unprecedented opportunity to obtain robust frequency estimates even for very rare variants.MethodsWe present a statistical framework for the frequency-based filtering of candidate disease-causing variants, accounting for disease prevalence, genetic and allelic heterogeneity, inheritance mode, penetrance, and sampling variance in reference datasets.ResultsUsing the example of cardiomyopathy, we show that our approach reduces by two-thirds the number of candidate variants under consideration in the average exome, without removing true pathogenic variants (false-positive rate<0.001).ConclusionWe outline a statistically robust framework for assessing whether a variant is "too common" to be causative for a Mendelian disorder of interest. We present precomputed allele frequency cutoffs for all variants in the ExAC dataset.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Bamshad MJ, Ng SB, Bigham AW et al, Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 2011;12:745–755. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
