

Genetically engineered probiotic for the treatment of phenylketonuria (PKU); assessment of a novel treatment in vitro and in the PAHenu2 mouse model of PKU
- PMID: 28520731
- PMCID: PMC5435137
- DOI: 10.1371/journal.pone.0176286
Genetically engineered probiotic for the treatment of phenylketonuria (PKU); assessment of a novel treatment in vitro and in the PAHenu2 mouse model of PKU
Abstract
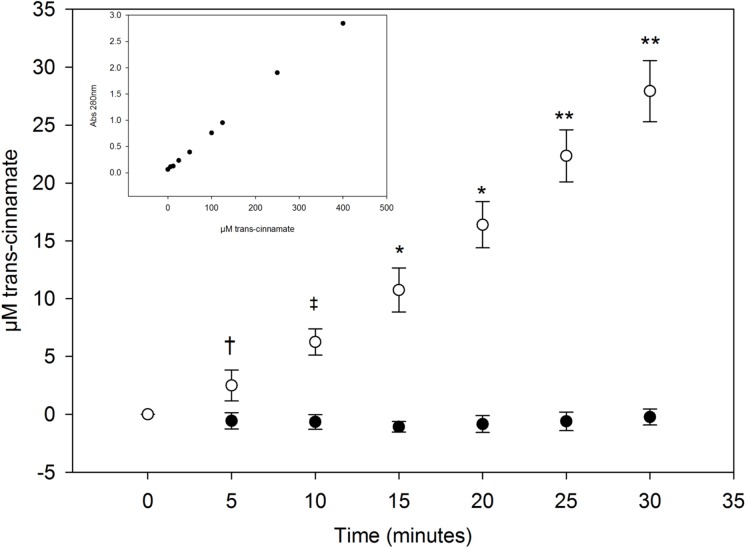
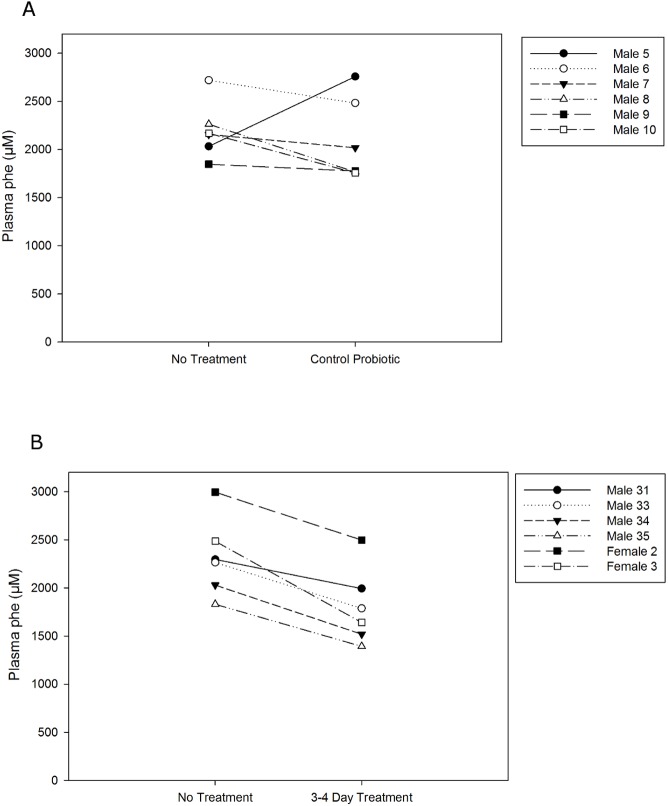
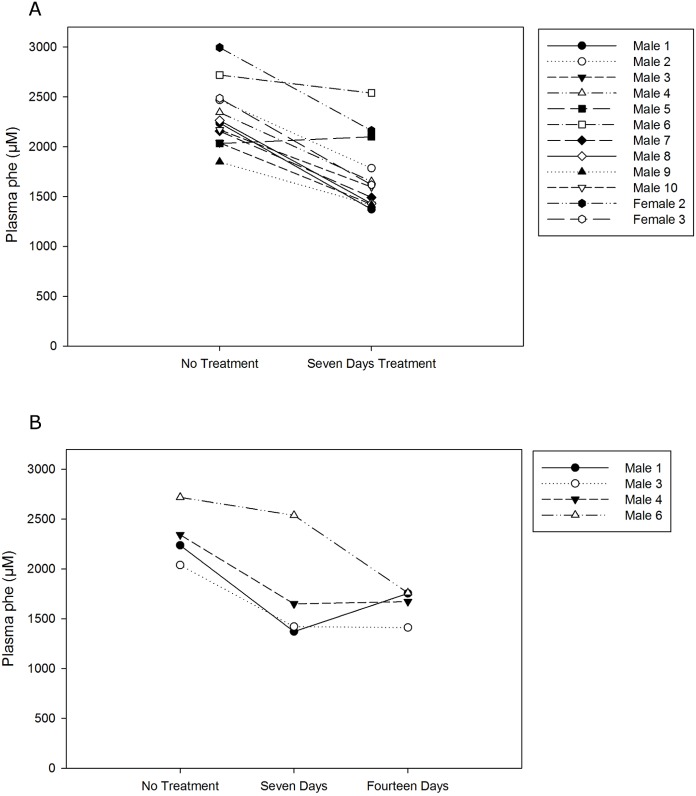
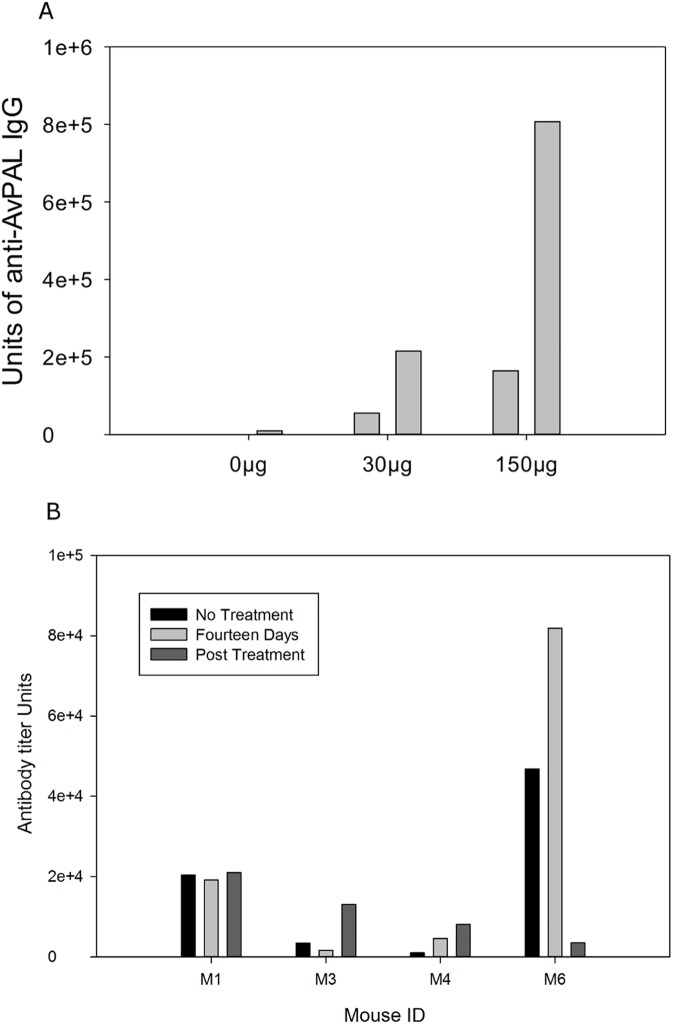
Phenylketonuria (PKU) is a genetic disease characterized by the inability to convert dietary phenylalanine to tyrosine by phenylalanine hydroxylase. Given the importance of gut microbes in digestion, a genetically engineered microbe could potentially degrade some ingested phenylalanine from the diet prior to absorption. To test this, a phenylalanine lyase gene from Anabaena variabilis (AvPAL) was codon-optimized and cloned into a shuttle vector for expression in Lactobacillus reuteri 100-23C (pHENOMMenal). Functional expression of AvPAL was determined in vitro, and subsequently tested in vivo in homozygous PAHenu2 (PKU model) mice. Initial trials of two PAHenu2 homozygous (PKU) mice defined conditions for freeze-drying and delivery of bacteria. Animals showed reduced blood phe within three to four days of treatment with pHENOMMenal probiotic, and blood phe concentrations remained significantly reduced (P < 0.0005) compared to untreated controls during the course of experiments. Although pHENOMMenal probiotic could be cultured from fecal samples at four months post treatment, it could no longer be cultivated from feces at eight months post treatment, indicating eventual loss of the microbe from the gut. Preliminary screens during experimentation found no immune response to AvPAL. Collectively these studies provide data for the use of a genetically engineered probiotic as a potential treatment for PKU.
Conflict of interest statement
Figures
References
-
- Brumm VL, Bilder D, Waisbren SE. Psychiatric symptoms and disorders in phenylketonuria. Molecular genetics and metabolism. 2010;99 Suppl 1:S59–63. Epub 2010/03/05. S1096-7192(09)00475-2 [pii]. - PubMed
-
- Hamman K, Clark H, Montini E, Al-Dhalimy M, Grompe M, Finegold M, et al. Low therapeutic threshold for hepatocyte replacement in murine phenylketonuria. Mol Ther. 2005;12(2):337–44. Epub 2005/07/27. PubMed Central PMCID: PMC2694052. doi: 10.1016/j.ymthe.2005.03.025 - DOI - PMC - PubMed
-
- Roato I, Porta F, Mussa A, D'Amico L, Fiore L, Garelli D, et al. Bone impairment in phenylketonuria is characterized by circulating osteoclast precursors and activated T cell increase. PLoS One. 2010;5(11):e14167 Epub 2010/12/15. PubMed Central PMCID: PMC2994752. doi: 10.1371/journal.pone.0014167 - DOI - PMC - PubMed
-
- Porta F, Roato I, Mussa A, Repici M, Gorassini E, Spada M, et al. Increased spontaneous osteoclastogenesis from peripheral blood mononuclear cells in phenylketonuria. J Inherit Metab Dis. 2008;31 Suppl 2:S339–42. Epub 2008/10/17. - PubMed
-
- McDonald JD, Dyer CA, Gailis L, Kirby ML. Cardiovascular defects among the progeny of mouse phenylketonuria females. Pediatr Res. 1997;42(1):103–7. Epub 1997/07/01. doi: 10.1203/00006450-199707000-00016 - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
