

Mass spectrometry for serine ADP-ribosylation? Think o-glycosylation!
- PMID: 28520971
- PMCID: PMC5499872
- DOI: 10.1093/nar/gkx446
Mass spectrometry for serine ADP-ribosylation? Think o-glycosylation!
Abstract
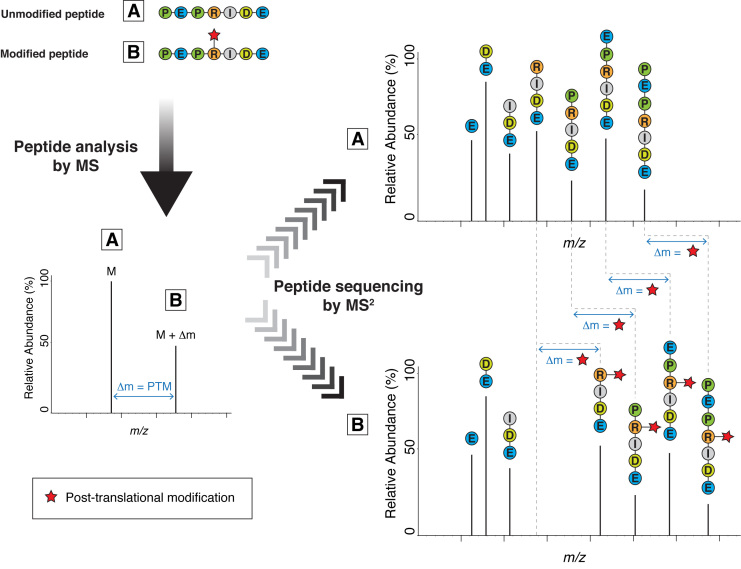
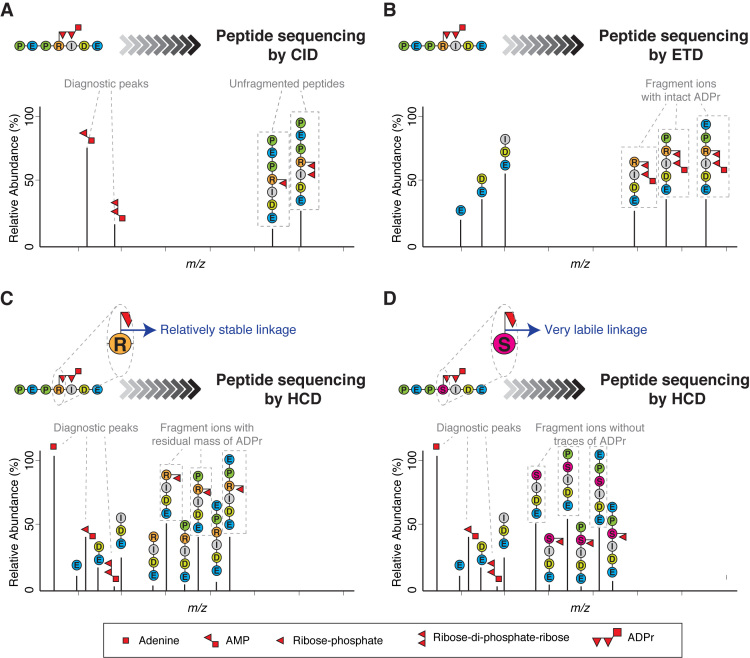
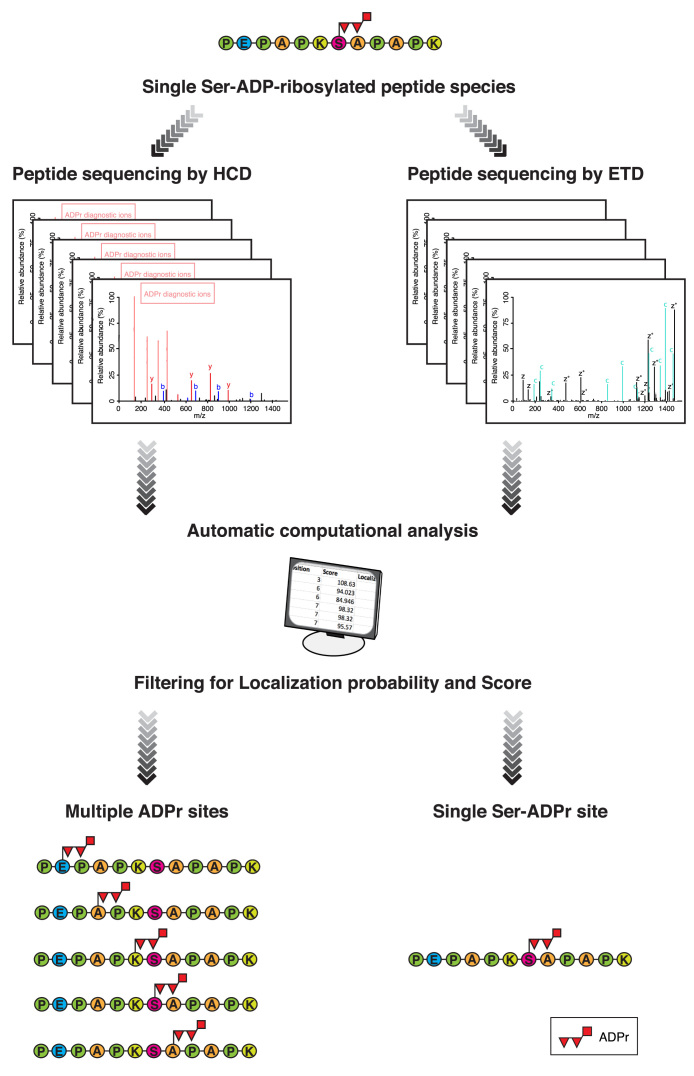
Protein ADP-ribosylation (ADPr), a biologically and clinically important post-translational modification, exerts its functions by targeting a variety of different amino acids. Its repertoire recently expanded to include serine ADPr, which is emerging as an important and widespread signal in the DNA damage response. Chemically, serine ADPr (and more generally o-glycosidic ADPr) is a form of o-glycosylation, and its extreme lability renders it practically invisible to standard mass spectrometry approaches, often leading to erroneous localizations. The knowledge from the mature field of o-glycosation and our own initial difficulties with mass spectrometric analyzes of serine ADPr suggest how to avoid these misidentifications and fully explore the scope of o-glycosidic ADPr in DNA damage response and beyond.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Hedberg C., Itzen A.. Molecular perspectives on protein adenylylation. Acs. Chem. Biol. 2015; 10:12–21. - PubMed
-
- Bock F.J., Chang P.. New directions in PARP biology. FEBS J. 2016; 283:4017–4031. - PubMed
-
- Martin-Hernandez K., Rodriguez-Vargas J.M., Schreiber V., Dantzer F.. Expanding functions of ADP-ribosylation in the maintenance of genome integrity. Semin. Cell Dev. Biol. 2017; 63:92–101. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
