

Drugging the Cancers Addicted to DNA Repair
- PMID: 28521333
- PMCID: PMC5436301
- DOI: 10.1093/jnci/djx059
Drugging the Cancers Addicted to DNA Repair
Abstract
Defects in DNA repair can result in oncogenic genomic instability. Cancers occurring from DNA repair defects were once thought to be limited to rare inherited mutations (such as BRCA1 or 2). It now appears that a clinically significant fraction of cancers have acquired DNA repair defects. DNA repair pathways operate in related networks, and cancers arising from loss of one DNA repair component typically become addicted to other repair pathways to survive and proliferate. Drug inhibition of the rescue repair pathway prevents the repair-deficient cancer cell from replicating, causing apoptosis (termed synthetic lethality). However, the selective pressure of inhibiting the rescue repair pathway can generate further mutations that confer resistance to the synthetic lethal drugs. Many such drugs currently in clinical use inhibit PARP1, a repair component to which cancers arising from inherited BRCA1 or 2 mutations become addicted. It is now clear that drugs inducing synthetic lethality may also be therapeutic in cancers with acquired DNA repair defects, which would markedly broaden their applicability beyond treatment of cancers with inherited DNA repair defects. Here we review how each DNA repair pathway can be attacked therapeutically and evaluate DNA repair components as potential drug targets to induce synthetic lethality. Clinical use of drugs targeting DNA repair will markedly increase when functional and genetic loss of repair components are consistently identified. In addition, future therapies will exploit artificial synthetic lethality, where complementary DNA repair pathways are targeted simultaneously in cancers without DNA repair defects.
© The Author 2017. Published by Oxford University Press.
Figures
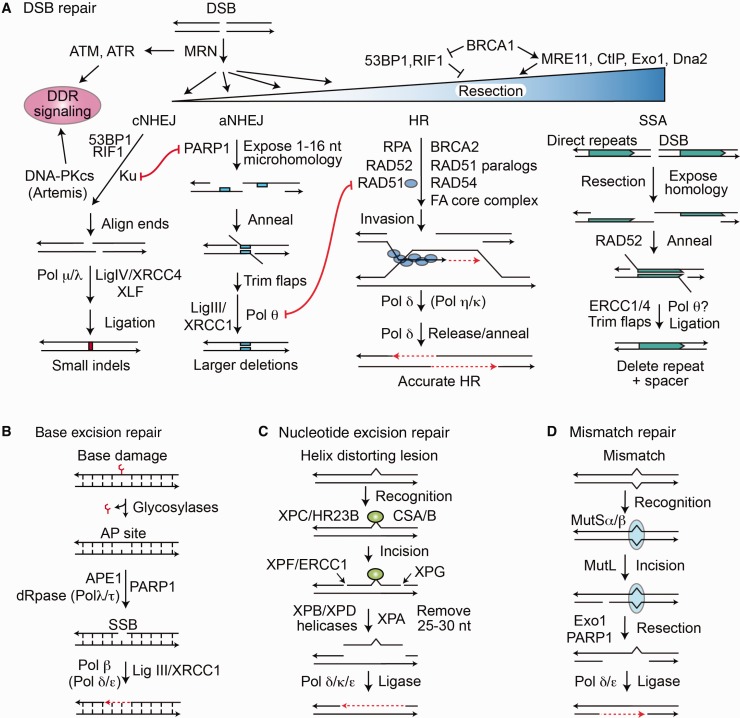
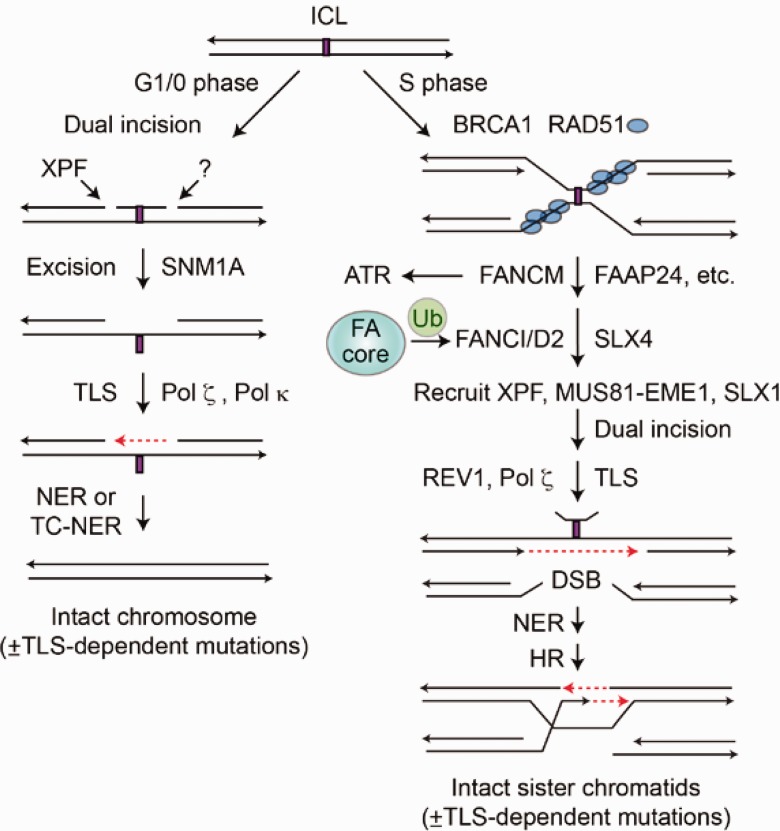
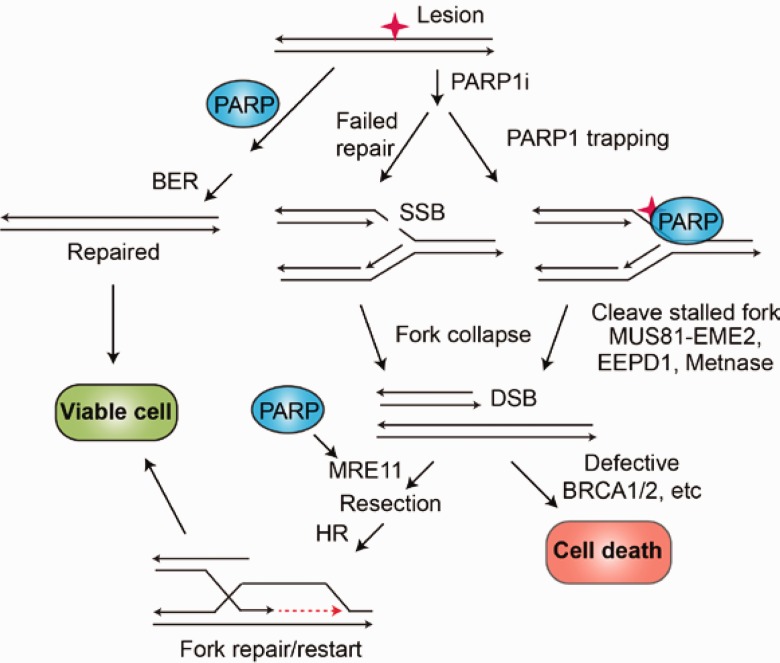
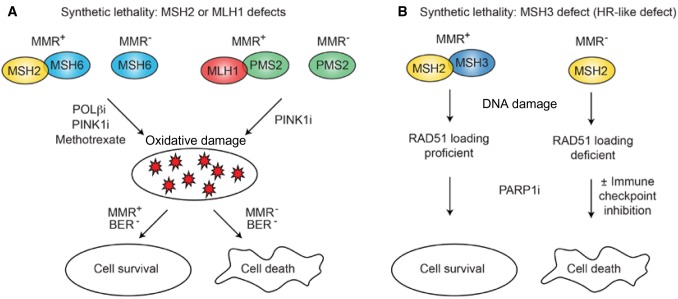
References
-
- Mazouzi A, Velimezi G, Loizou JI.. DNA replication stress: Causes, resolution and disease. Exp Cell Res. 2014;329(1):85–93. - PubMed
-
- Dizdaroglu M. Oxidatively induced DNA damage and its repair in cancer. Mutat Res Rev Mutat Res. 2015;763:212–245. - PubMed
-
- Hanks S, Coleman K, Reid S, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 2004;36:1159–1161. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
