

Polycystic Kidney Disease without an Apparent Family History
- PMID: 28522688
- PMCID: PMC5576926
- DOI: 10.1681/ASN.2016090938
Polycystic Kidney Disease without an Apparent Family History
Abstract
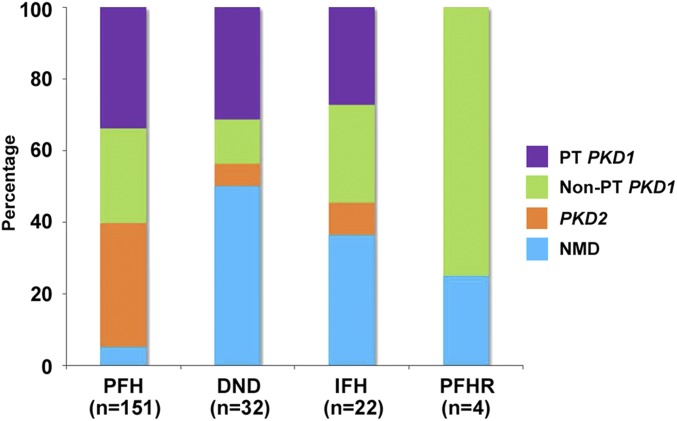
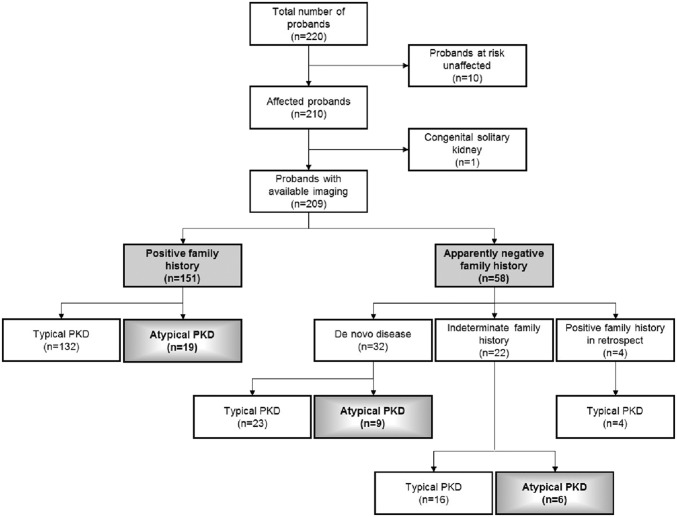
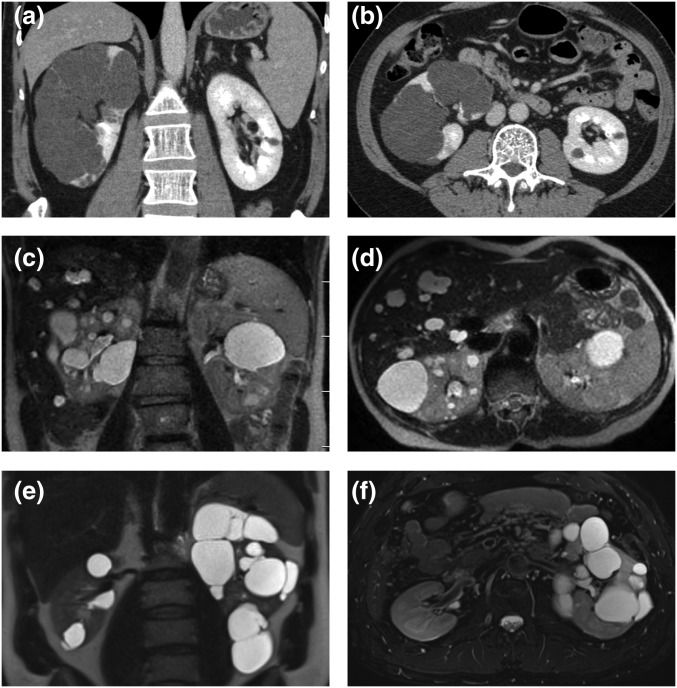
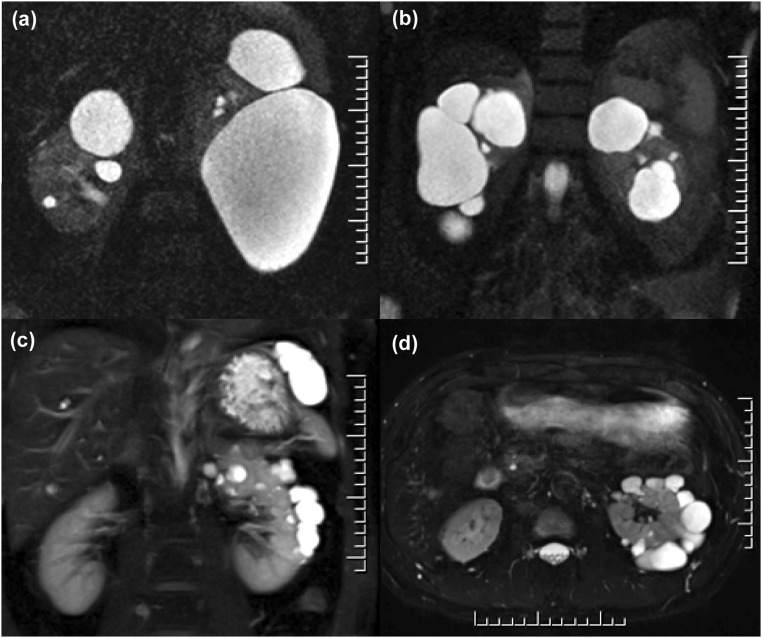
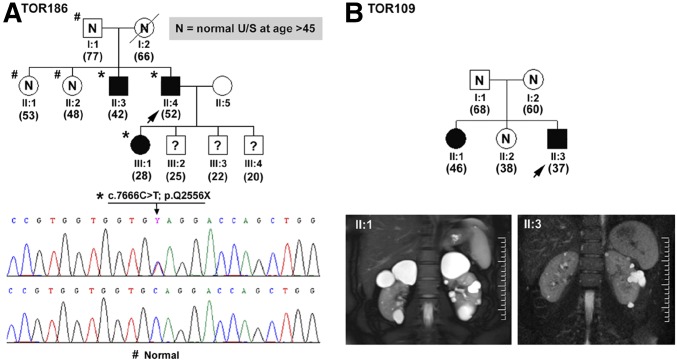
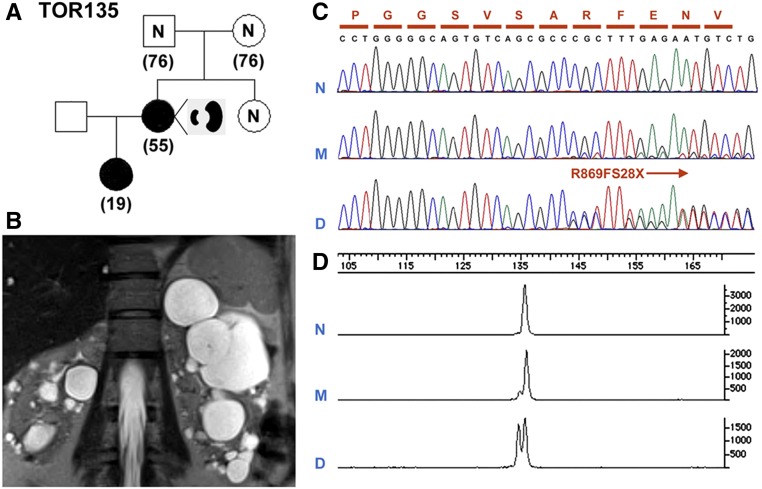
The absence of a positive family history (PFH) in 10%-25% of patients poses a diagnostic challenge for autosomal dominant polycystic kidney disease (ADPKD). In the Toronto Genetic Epidemiology Study of Polycystic Kidney Disease, 210 affected probands underwent renal function testing, abdominal imaging, and comprehensive PKD1 and PKD2 mutation screening. From this cohort, we reviewed all patients with and without an apparent family history, examined their parental medical records, and performed renal imaging in all available parents of unknown disease status. Subsequent reclassification of 209 analyzed patients revealed 72.2% (151 of 209) with a PFH, 15.3% (32 of 209) with de novo disease, 10.5% (22 of 209) with an indeterminate family history, and 1.9% (four of 209) with PFH in retrospect. Among the patients with de novo cases, we found two families with germline mosaicism and one family with somatic mosaicism. Additionally, analysis of renal imaging revealed that 16.3% (34 of 209) of patients displayed atypical PKD, most of which followed one of three patterns: asymmetric or focal PKD with PFH and an identified PKD1 or PKD2 mutation (15 of 34), asymmetric and de novo PKD with proven or suspected somatic mosaicism (seven of 34), or focal PKD without any identifiable PKD1 or PKD2 mutation (eight of 34). In conclusion, PKD without an apparent family history may be due to de novo disease, missing parental medical records, germline or somatic mosaicism, or mild disease from hypomorphic PKD1 and PKD2 mutations. Furthermore, mutations of a newly identified gene for ADPKD, GANAB, and somatic mosaicism need to be considered in the mutation-negative patients with focal disease.
Keywords: family history; human genetics; polycystic kidney disease.
Copyright © 2017 by the American Society of Nephrology.
Figures
References
-
- Barua M, Pei Y: Diagnosis of autosomal-dominant polycystic kidney disease: An integrated approach. Semin Nephrol 30: 356–365, 2010 - PubMed
-
- Grantham JJ, Mulamalla S, Swenson-Fields KI: Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol 7: 556–566, 2011 - PubMed
-
- Heyer CM, Sundsbak JL, Abebe KZ, Chapman AB, Torres VE, Grantham JJ, Bae KT, Schrier RW, Perrone RD, Braun WE, Steinman TI, Mrug M, Yu AS, Brosnahan G, Hopp K, Irazabal MV, Bennett WM, Flessner MF, Moore CG, Landsittel D, Harris PC; HALT PKD and CRISP Investigators : Predicted mutation strength of non-truncating PKD1 mutations aids genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 2872–2884, 2016 - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
