

Rubinstein-Taybi Syndrome and Epigenetic Alterations
- PMID: 28523540
- PMCID: PMC6863608
- DOI: 10.1007/978-3-319-53889-1_3
Rubinstein-Taybi Syndrome and Epigenetic Alterations
Abstract
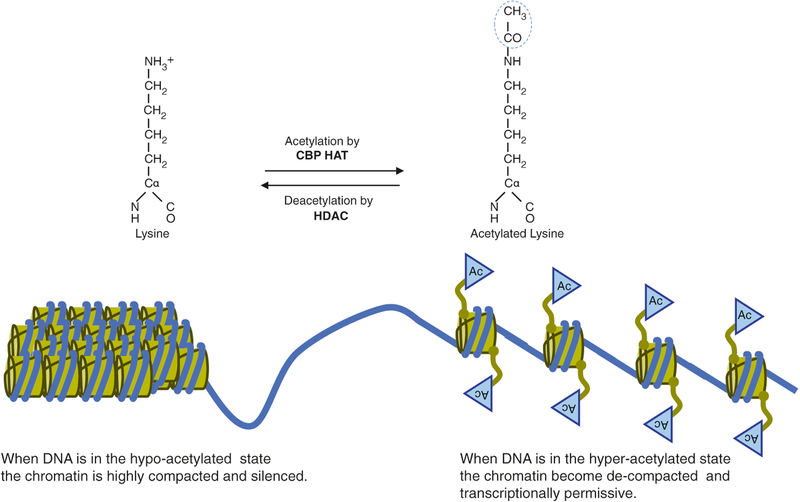
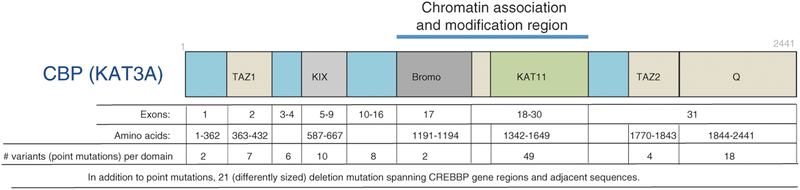
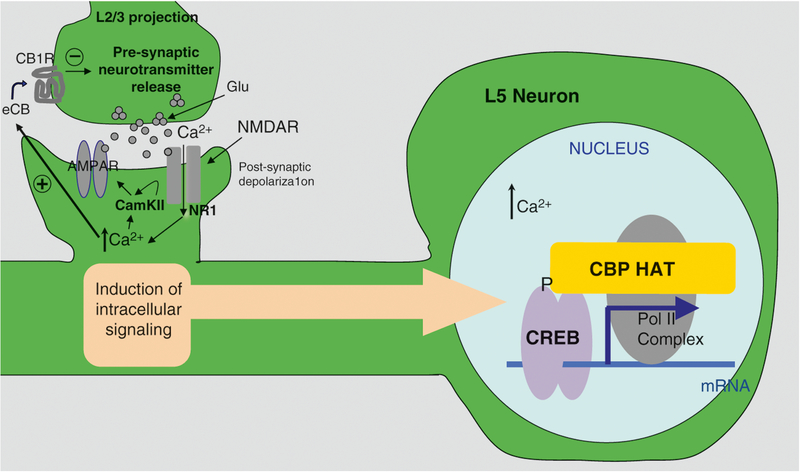
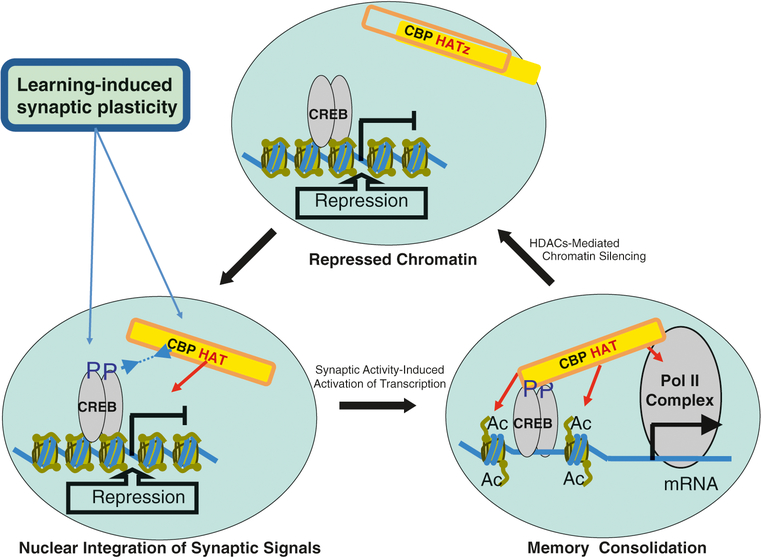
Rubinstein-Taybi syndrome (RSTS) is a rare genetic disorder in humans characterized by growth and psychomotor delay, abnormal gross anatomy, and mild to severe mental retardation (Rubinstein and Taybi, Am J Dis Child 105:588-608, 1963, Hennekam et al., Am J Med Genet Suppl 6:56-64, 1990). RSTS is caused by de novo mutations in epigenetics-associated genes, including the cAMP response element-binding protein (CREBBP), the gene-encoding protein referred to as CBP, and the EP300 gene, which encodes the p300 protein, a CBP homologue. Recent studies of the epigenetic mechanisms underlying cognitive functions in mice provide direct evidence for the involvement of nuclear factors (e.g., CBP) in the control of higher cognitive functions. In fact, a role for CBP in higher cognitive function is suggested by the finding that RSTS is caused by heterozygous mutations at the CBP locus (Petrij et al., Nature 376:348-351, 1995). CBP was demonstrated to possess an intrinsic histone acetyltransferase activity (Ogryzko et al., Cell 87:953-959, 1996) that is required for CREB-mediated gene expression (Korzus et al., Science 279:703-707, 1998). The intrinsic protein acetyltransferase activity in CBP might directly destabilize promoter-bound nucleosomes, facilitating the activation of transcription. Due to the complexity of developmental abnormalities and the possible genetic compensation associated with this congenital disorder, however, it is difficult to establish a direct role for CBP in cognitive function in the adult brain. Although aspects of the clinical presentation in RSTS cases have been extensively studied, a spectrum of symptoms found in RSTS patients can be accessed only after birth, and, thus, prenatal genetic tests for this extremely rare genetic disorder are seldom considered. Even though there has been intensive research on the genetic and epigenetic function of the CREBBP gene in rodents, the etiology of this devastating congenital human disorder is largely unknown.
Keywords: CBP; CREBBP; EP300; Epigenetic; HDAC; Histone acetylation; Memory; RSTS; Rubinstein-Taybi syndrome; p300.
Figures


References
-
- Rosenfeld MG, Glass CK. Coregulator codes of transcriptional regulation by nuclear receptors. J Biol Chem. 2001;276(40):36865–8. - PubMed
-
- Neely KE, Workman JL. Histone acetylation and chromatin remodeling: which comes first? Mol Genet Metab. 2002;76(1):1–5. - PubMed
-
- Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell Biol. 2003;15(2):172–83. - PubMed
-
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365(6449):855–9. - PubMed
-
- Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, Livingston DM. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994;8(8):869–84. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
