

The golden retriever model of Duchenne muscular dystrophy
- PMID: 28526070
- PMCID: PMC5438519
- DOI: 10.1186/s13395-017-0124-z
The golden retriever model of Duchenne muscular dystrophy
Abstract
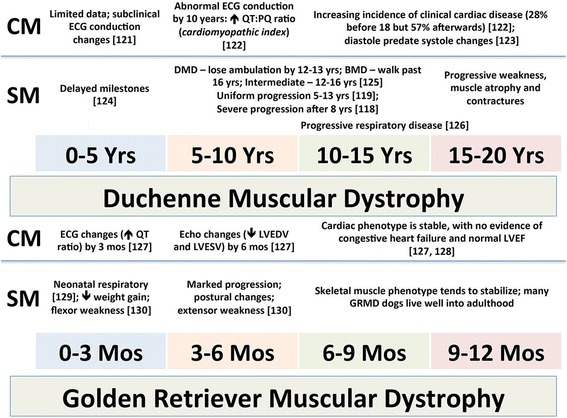
Duchenne muscular dystrophy (DMD) is an X-linked disease caused by mutations in the DMD gene and loss of the protein dystrophin. The absence of dystrophin leads to myofiber membrane fragility and necrosis, with eventual muscle atrophy and contractures. Affected boys typically die in their second or third decade due to either respiratory failure or cardiomyopathy. Despite extensive attempts to develop definitive therapies for DMD, the standard of care remains prednisone, which has only palliative benefits. Animal models, mainly the mdx mouse and golden retriever muscular dystrophy (GRMD) dog, have played a key role in studies of DMD pathogenesis and treatment development. Because the GRMD clinical syndrome is more severe than in mice, better aligning with the progressive course of DMD, canine studies may translate better to humans. The original founder dog for all GRMD colonies worldwide was identified in the early 1980s before the discovery of the DMD gene and dystrophin. Accordingly, analogies to DMD were initially drawn based on similar clinical features, ranging from the X-linked pattern of inheritance to overlapping histopathologic lesions. Confirmation of genetic homology between DMD and GRMD came with identification of the underlying GRMD mutation, a single nucleotide change that leads to exon skipping and an out-of-frame DMD transcript. GRMD colonies have subsequently been established to conduct pathogenetic and preclinical treatment studies. Simultaneous with the onset of GRMD treatment trials, phenotypic biomarkers were developed, allowing definitive characterization of treatment effect. Importantly, GRMD studies have not always substantiated findings from mdx mice and have sometimes identified serious treatment side effects. While the GRMD model may be more clinically relevant than the mdx mouse, usage has been limited by practical considerations related to expense and the number of dogs available. This further complicates ongoing broader concerns about the poor rate of translation of animal model preclinical studies to humans with analogous diseases. Accordingly, in performing GRMD trials, special attention must be paid to experimental design to align with the approach used in DMD clinical trials. This review provides context for the GRMD model, beginning with its original description and extending to its use in preclinical trials.
Keywords: Animal models; Duchenne muscular dystrophy (DMD); Golden retriever muscular dystrophy (GRMD); Preclinical studies.
Figures
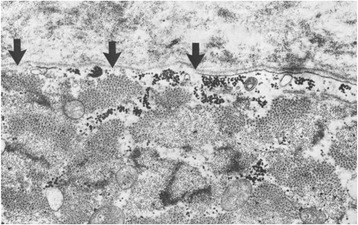
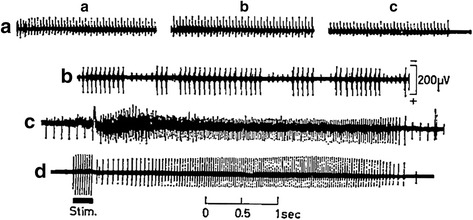

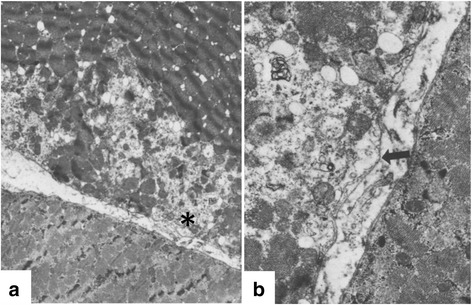
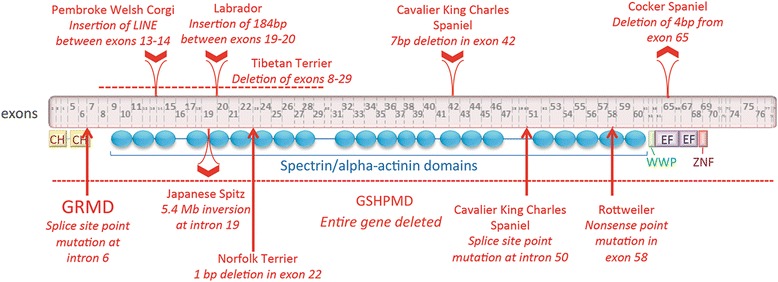
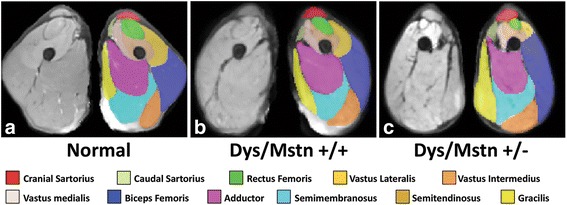
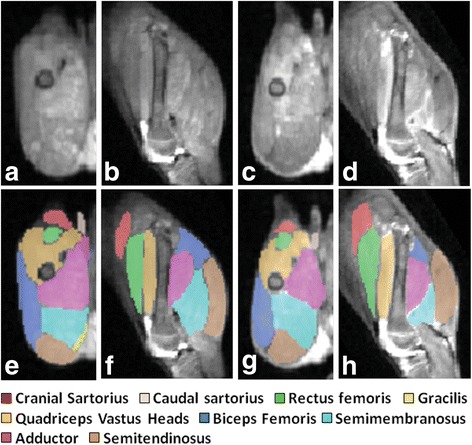
References
-
- Multiple authors Experimental primary myopathies and their relationship to human disease. Ann NY Acad Sci. 1966;138:3–366.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
