

Role of GPER in estrogen-dependent nitric oxide formation and vasodilation
- PMID: 28529128
- PMCID: PMC5694388
- DOI: 10.1016/j.jsbmb.2017.05.006
Role of GPER in estrogen-dependent nitric oxide formation and vasodilation
Abstract
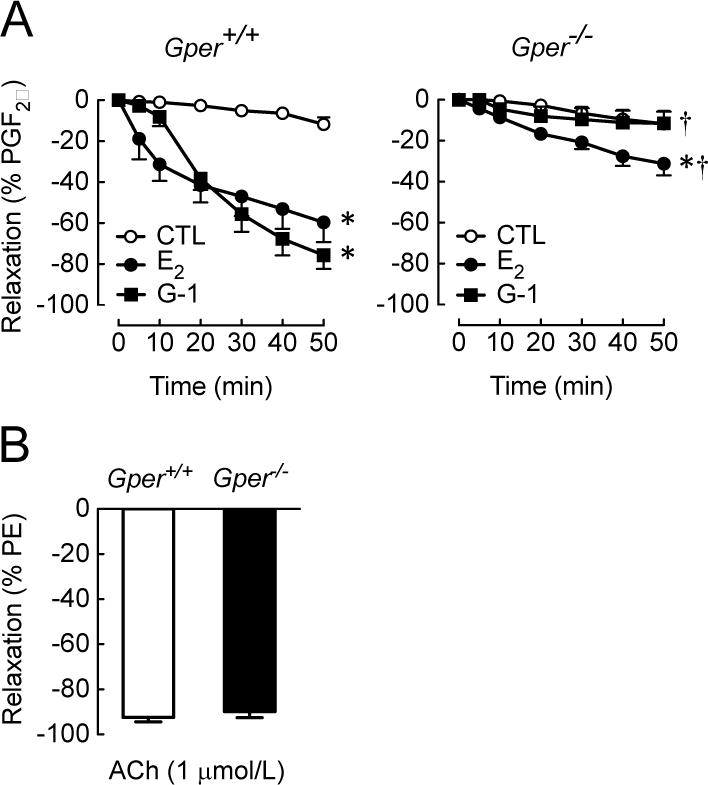
Estrogens are potent regulators of vasomotor tone, yet underlying receptor- and ligand-specific signaling pathways remain poorly characterized. The primary physiological estrogen 17β-estradiol (E2), a non-selective agonist of classical nuclear estrogen receptors (ERα and ERβ) as well as the G protein-coupled estrogen receptor (GPER), stimulates formation of the vasodilator nitric oxide (NO) in endothelial cells. Here, we studied the contribution of GPER signaling in E2-dependent activation of endothelial NO formation and subsequent vasodilation. Employing E2 and the GPER-selective agonist G-1, we investigated eNOS phosphorylation and NO formation in human endothelial cells, and endothelium-dependent vasodilation in the aortae of wild-type and Gper-deficient mice. Both E2 and G-1 induced phosphorylation of eNOS at the activation site Ser1177 to similar extents. Endothelial NO production to E2 was comparable to that of G-1, and was substantially reduced after pharmacological inhibition of GPER. Similarly, the clinically used ER-targeting drugs 4OH-tamoxifen, raloxifene, and ICI182,780 (faslodex, fulvestrant™) induced NO formation in part via GPER. We identified c-Src, EGFR, PI3K and ERK signaling pathways to be involved in GPER-dependent NO formation. In line with activation of NO formation in cells, E2 and G-1 induced equally potent vasodilation in the aorta of wild-type mice. Gper deletion completely abrogated the vasodilator response to G-1, while reducing the response to E2 by ∼50%. These findings indicate that a substantial portion of E2-induced endothelium-dependent vasodilation and NO formation is mediated by GPER. Thus, selective targeting of vascular GPER may be a suitable approach to activate the endothelial NO pathway, possibly leading to reduced vasomotor tone and inhibition of atherosclerotic vascular disease.
Keywords: Endothelium; Estrogen; GPER; GPR30; NO; SERD; SERM; Vascular; Vasodilation; eNOS.
Copyright © 2017 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Adaptive increases in expression and vasodilator activity of estrogen receptor subtypes in a blood vessel-specific pattern during pregnancy.Am J Physiol Heart Circ Physiol. 2015 Nov 15;309(10):H1679-96. doi: 10.1152/ajpheart.00532.2015. Epub 2015 Sep 25. Am J Physiol Heart Circ Physiol. 2015. PMID: 26408543 Free PMC article.
-
Estrogen Enhances Linkage in the Vascular Endothelial Calmodulin Network via a Feedforward Mechanism at the G Protein-coupled Estrogen Receptor 1.J Biol Chem. 2016 May 13;291(20):10805-23. doi: 10.1074/jbc.M115.697334. Epub 2016 Mar 17. J Biol Chem. 2016. PMID: 26987903 Free PMC article.
-
17β-Estradiol and Agonism of G-protein-Coupled Estrogen Receptor Enhance Hippocampal Memory via Different Cell-Signaling Mechanisms.J Neurosci. 2016 Mar 16;36(11):3309-21. doi: 10.1523/JNEUROSCI.0257-15.2016. J Neurosci. 2016. PMID: 26985039 Free PMC article.
-
Role of G-protein-coupled estrogen receptor (GPER/GPR30) in maintenance of meiotic arrest in fish oocytes.J Steroid Biochem Mol Biol. 2017 Mar;167:153-161. doi: 10.1016/j.jsbmb.2016.12.005. Epub 2016 Dec 19. J Steroid Biochem Mol Biol. 2017. PMID: 28007532 Review.
-
The G protein-coupled estrogen receptor GPER/GPR30 as a regulator of cardiovascular function.Vascul Pharmacol. 2011 Jul-Sep;55(1-3):17-25. doi: 10.1016/j.vph.2011.06.003. Epub 2011 Jul 5. Vascul Pharmacol. 2011. PMID: 21742056 Free PMC article. Review.
Cited by
-
G protein-coupled estrogen receptor 1 as a novel regulator of blood pressure.Am J Physiol Renal Physiol. 2020 Oct 1;319(4):F612-F617. doi: 10.1152/ajprenal.00045.2020. Epub 2020 Sep 7. Am J Physiol Renal Physiol. 2020. PMID: 32893662 Free PMC article. Review.
-
G Protein-Coupled Estrogen Receptor GPER: Molecular Pharmacology and Therapeutic Applications.Annu Rev Pharmacol Toxicol. 2023 Jan 20;63:295-320. doi: 10.1146/annurev-pharmtox-031122-121944. Annu Rev Pharmacol Toxicol. 2023. PMID: 36662583 Free PMC article. Review.
-
Female mice are protected from impaired parenchymal arteriolar TRPV4 function and impaired cognition in hypertension.Am J Physiol Heart Circ Physiol. 2023 May 1;324(5):H581-H597. doi: 10.1152/ajpheart.00481.2022. Epub 2023 Mar 10. Am J Physiol Heart Circ Physiol. 2023. PMID: 36897751 Free PMC article.
-
G-Protein-Coupled Estrogen Receptor Expression in Rat Uterine Artery Is Increased by Pregnancy and Induces Dilation in a Ca2+ and ERK1/2 Dependent Manner.Int J Mol Sci. 2022 May 26;23(11):5996. doi: 10.3390/ijms23115996. Int J Mol Sci. 2022. PMID: 35682675 Free PMC article.
-
Sex differences in vascular aging and impact of GPER deletion.Am J Physiol Heart Circ Physiol. 2022 Aug 1;323(2):H336-H349. doi: 10.1152/ajpheart.00238.2022. Epub 2022 Jun 24. Am J Physiol Heart Circ Physiol. 2022. PMID: 35749718 Free PMC article.
References
-
- Schenck-Gustafsson K, Brincat M, Erel CT, Gambacciani M, Lambrinoudaki I, Moen MH, Tremollieres F, Vujovic S, Rozenberg S, Rees M, Emas EMAS position statement: Managing the menopause in the context of coronary heart disease. Maturitas. 2011;68:94–7. - PubMed
-
- Meyer MR, Barton M. Estrogens and Coronary Artery Disease: New Clinical Perspectives. Adv Pharmacol. 2016;77:307–60. - PubMed
-
- Holm A, Nilsson BO. Identification and characterization of new mechanisms in vascular oestrogen signalling. Basic Clin Pharmacol Toxicol. 2013;113:287–93. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
