

Intestinal fungi contribute to development of alcoholic liver disease
- PMID: 28530644
- PMCID: PMC5490775
- DOI: 10.1172/JCI90562
Intestinal fungi contribute to development of alcoholic liver disease
Abstract
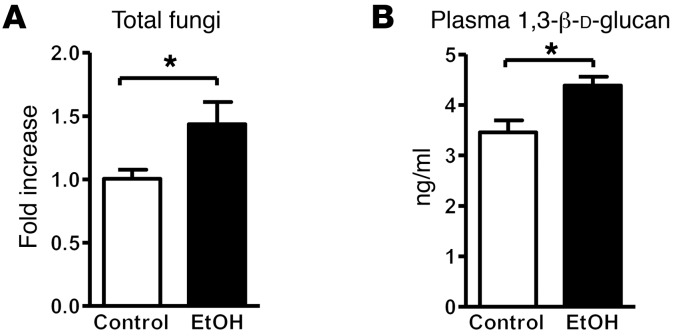
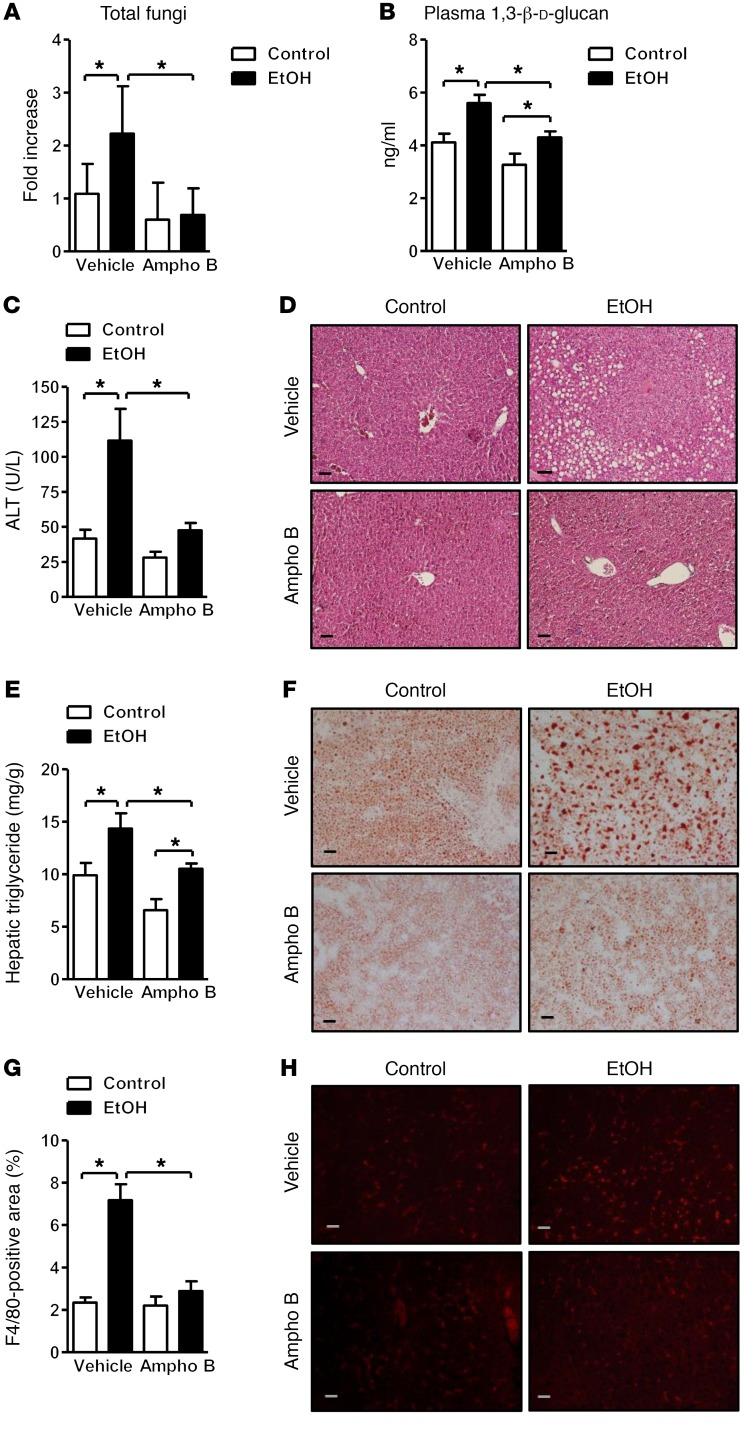
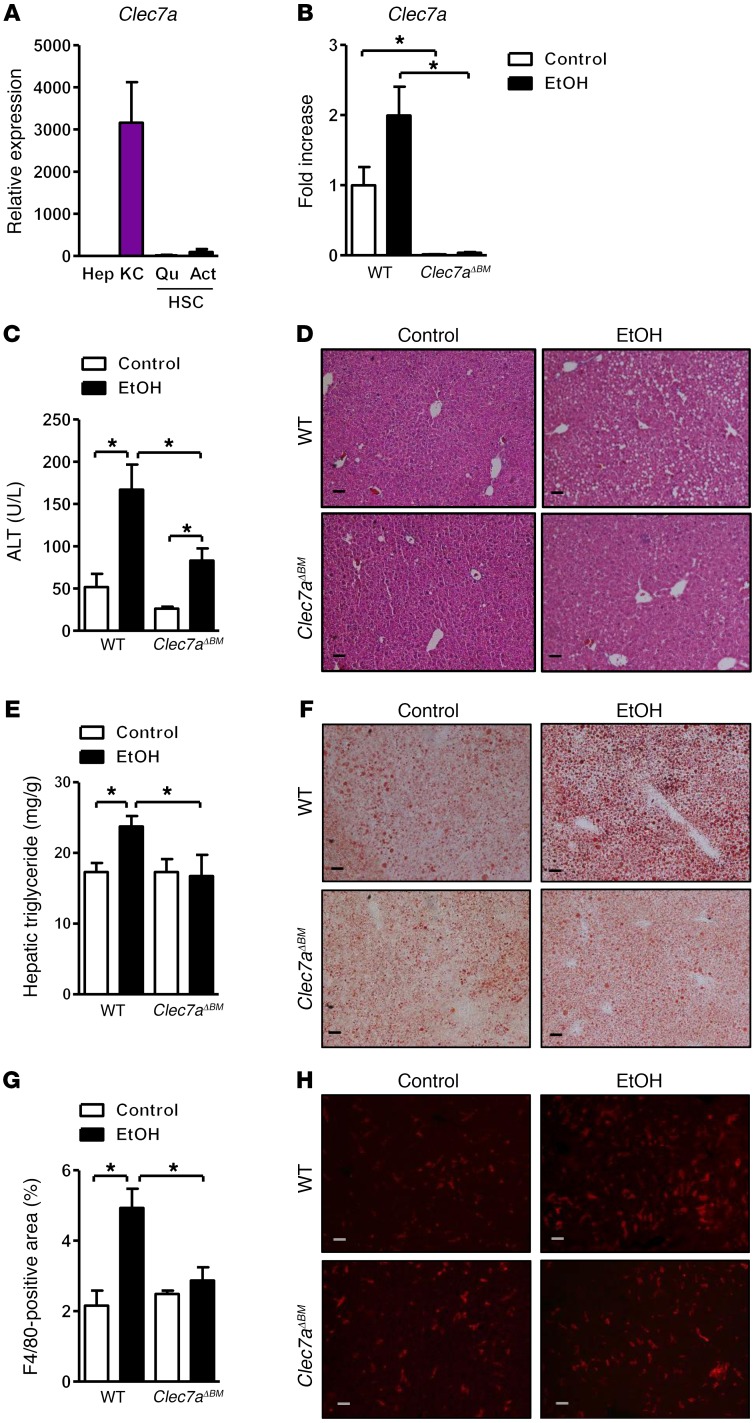
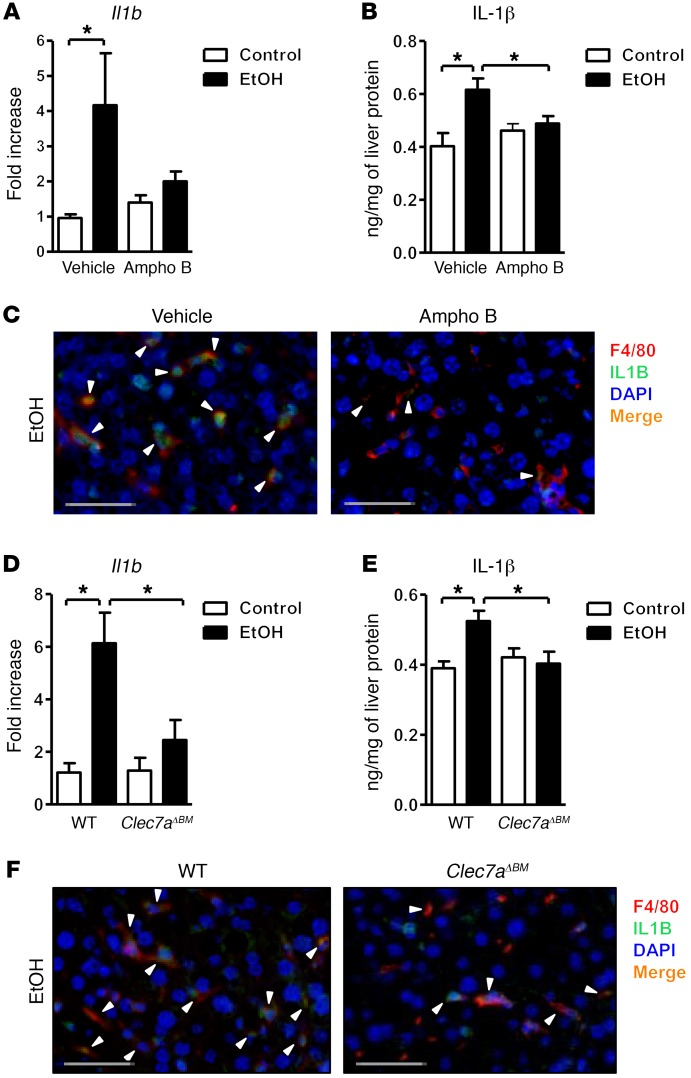
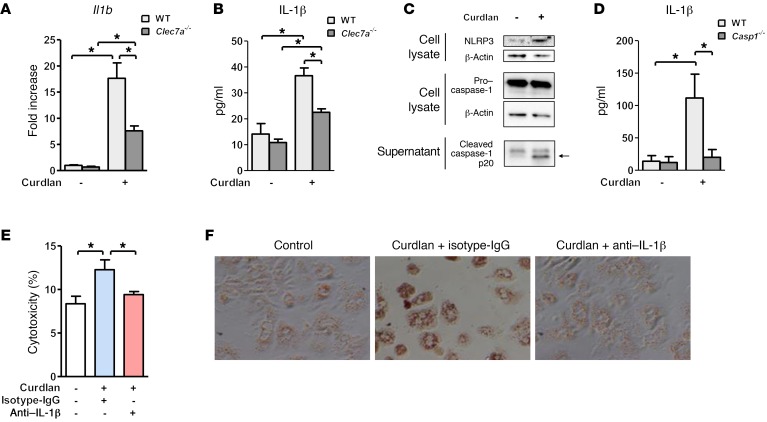
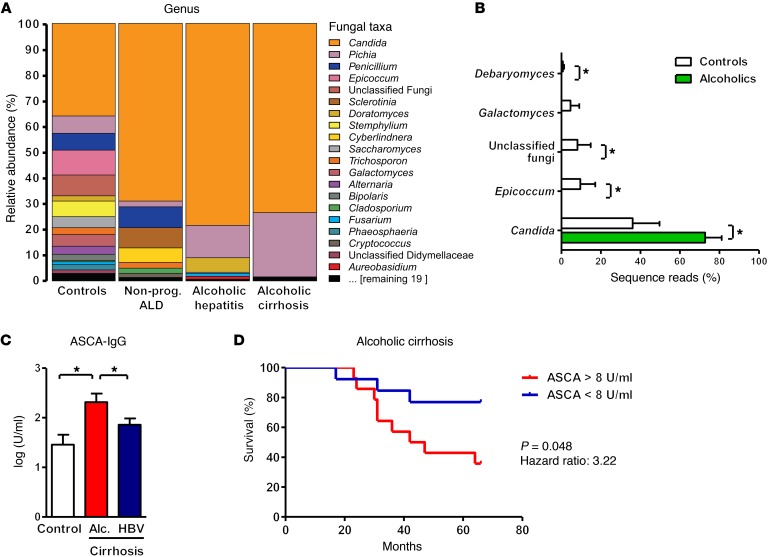
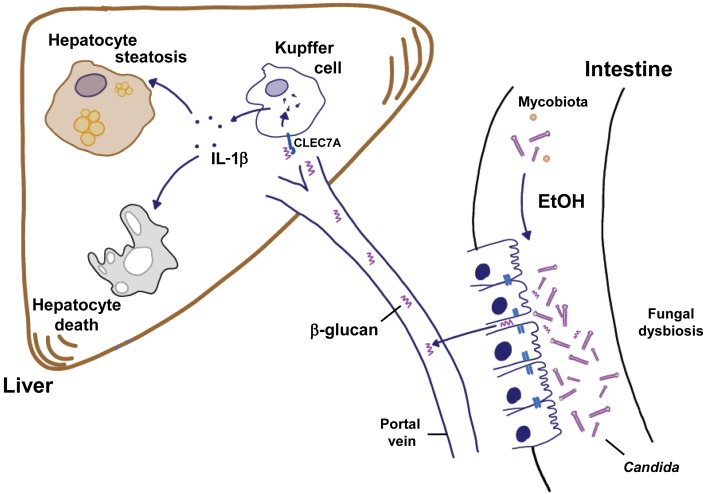
Chronic liver disease with cirrhosis is the 12th leading cause of death in the United States, and alcoholic liver disease accounts for approximately half of all cirrhosis deaths. Chronic alcohol consumption is associated with intestinal bacterial dysbiosis, yet we understand little about the contribution of intestinal fungi, or mycobiota, to alcoholic liver disease. Here we have demonstrated that chronic alcohol administration increases mycobiota populations and translocation of fungal β-glucan into systemic circulation in mice. Treating mice with antifungal agents reduced intestinal fungal overgrowth, decreased β-glucan translocation, and ameliorated ethanol-induced liver disease. Using bone marrow chimeric mice, we found that β-glucan induces liver inflammation via the C-type lectin-like receptor CLEC7A on Kupffer cells and possibly other bone marrow-derived cells. Subsequent increases in IL-1β expression and secretion contributed to hepatocyte damage and promoted development of ethanol-induced liver disease. We observed that alcohol-dependent patients displayed reduced intestinal fungal diversity and Candida overgrowth. Compared with healthy individuals and patients with non-alcohol-related cirrhosis, alcoholic cirrhosis patients had increased systemic exposure and immune response to mycobiota. Moreover, the levels of extraintestinal exposure and immune response correlated with mortality. Thus, chronic alcohol consumption is associated with an altered mycobiota and translocation of fungal products. Manipulating the intestinal mycobiome might be an effective strategy for attenuating alcohol-related liver disease.
Conflict of interest statement
Figures
Comment in
-
Gut microbiota: Intestinal fungi fuel the inflammatory fire in alcoholic liver disease.Nat Rev Gastroenterol Hepatol. 2017 Jul;14(7):385. doi: 10.1038/nrgastro.2017.78. Epub 2017 Jun 7. Nat Rev Gastroenterol Hepatol. 2017. PMID: 28588263 No abstract available.
-
Gut-Liver Axis Beyond the Microbiome: How the Fungal Mycobiome Contributes to Alcoholic Liver Disease.Hepatology. 2018 Dec;68(6):2426-2428. doi: 10.1002/hep.30055. Epub 2018 Jul 10. Hepatology. 2018. PMID: 29684245 No abstract available.
References
-
- WHO. Global Status Report On Alcohol And Health 2014. World Health Organization website. http://www.who.int/substance_abuse/publications/global_alcohol_report/en/ Accessed April 10, 2017.
-
- Bode JC, Bode C, Heidelbach R, Dürr HK, Martini GA. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology. 1984;31(1):30–34. - PubMed
MeSH terms
Substances
Grants and funding
- R01 AA020703/AA/NIAAA NIH HHS/United States
- U01 AA021912/AA/NIAAA NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- U01 AA021908/AA/NIAAA NIH HHS/United States
- U01 AA021856/AA/NIAAA NIH HHS/United States
- R01 DK099205/DK/NIDDK NIH HHS/United States
- I01 BX003259/BX/BLRD VA/United States
- R01 AA023417/AA/NIAAA NIH HHS/United States
- I01 BX002213/BX/BLRD VA/United States
- P30 DK034989/DK/NIDDK NIH HHS/United States
- R01 AA024726/AA/NIAAA NIH HHS/United States
- MR/N006364/1/MRC_/Medical Research Council/United Kingdom
- U01 AA024726/AA/NIAAA NIH HHS/United States
- RC2 AA019405/AA/NIAAA NIH HHS/United States
- 102705/Z/13/Z/WT_/Wellcome Trust/United Kingdom
- R01 AA020216/AA/NIAAA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
