

Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease
- PMID: 28530676
- PMCID: PMC5687889
- DOI: 10.1038/ng.3871
Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease
Abstract
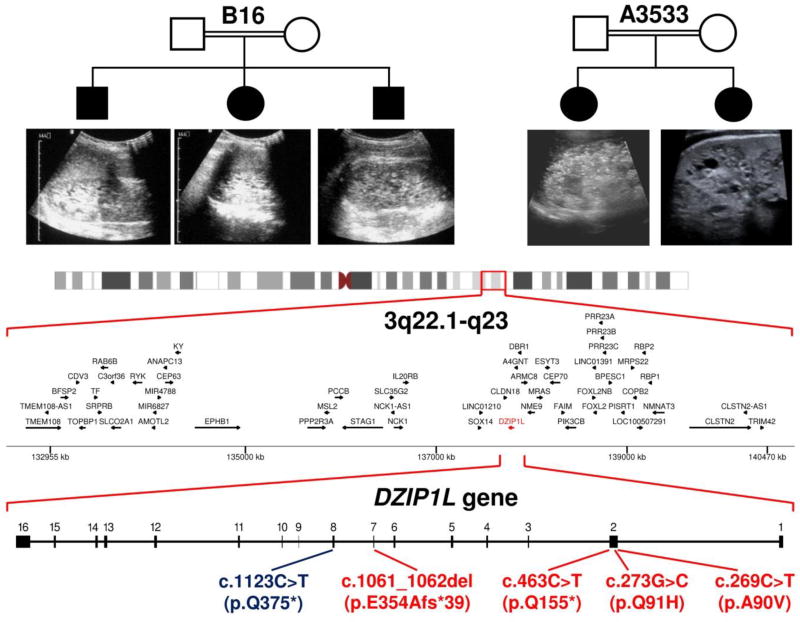
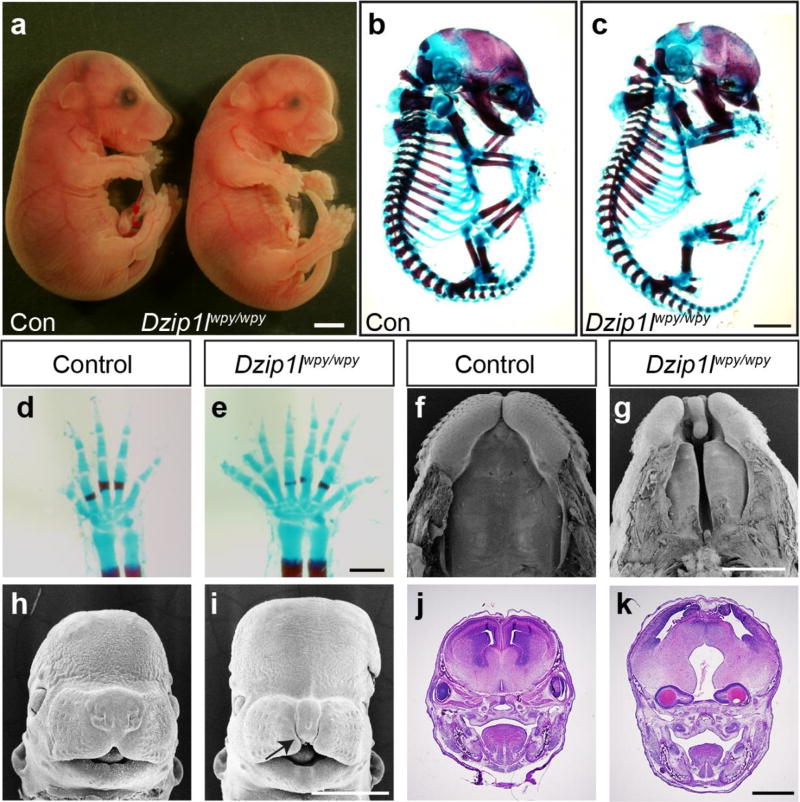
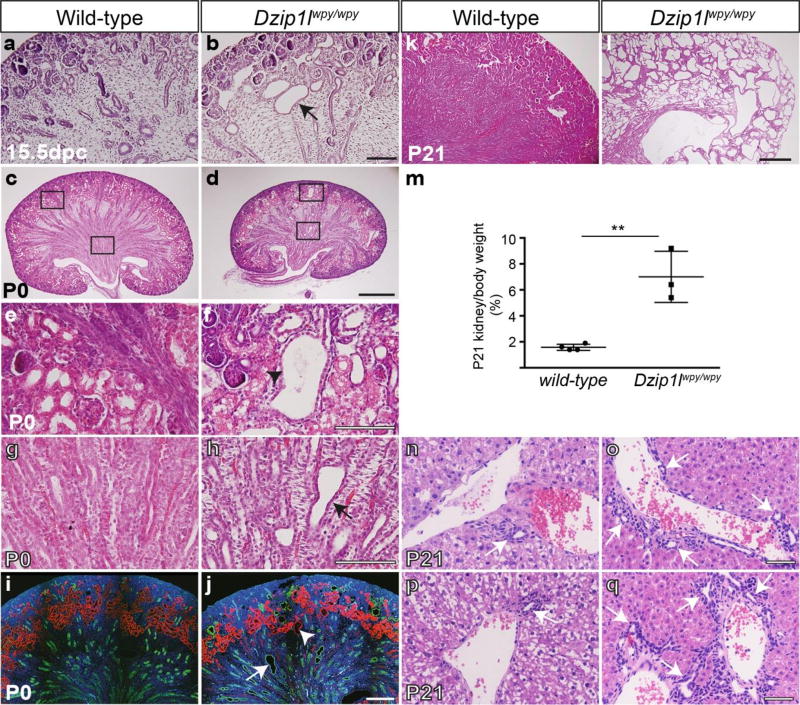
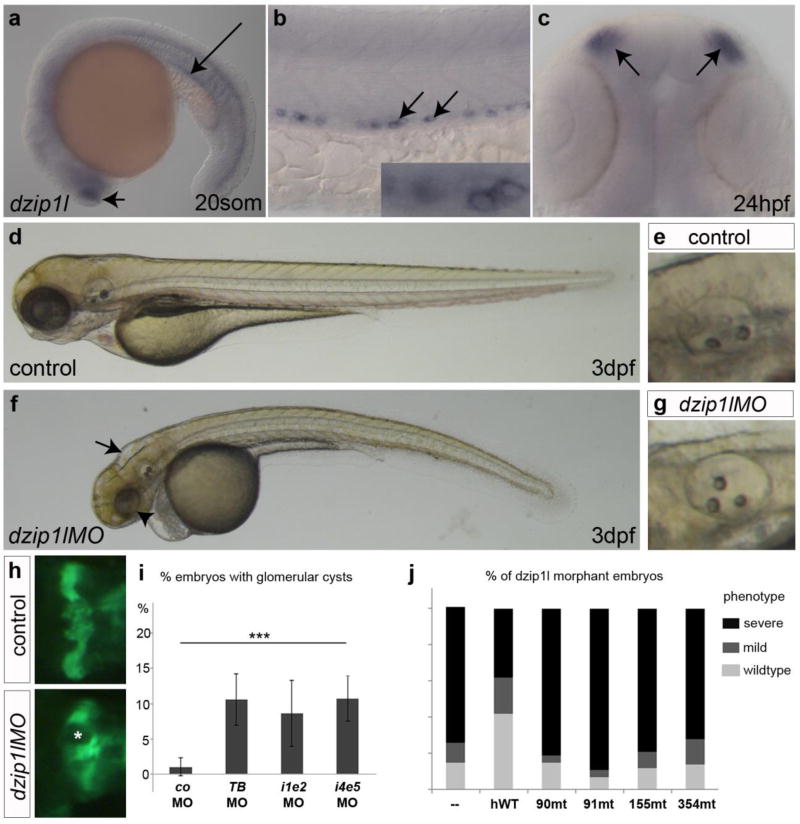
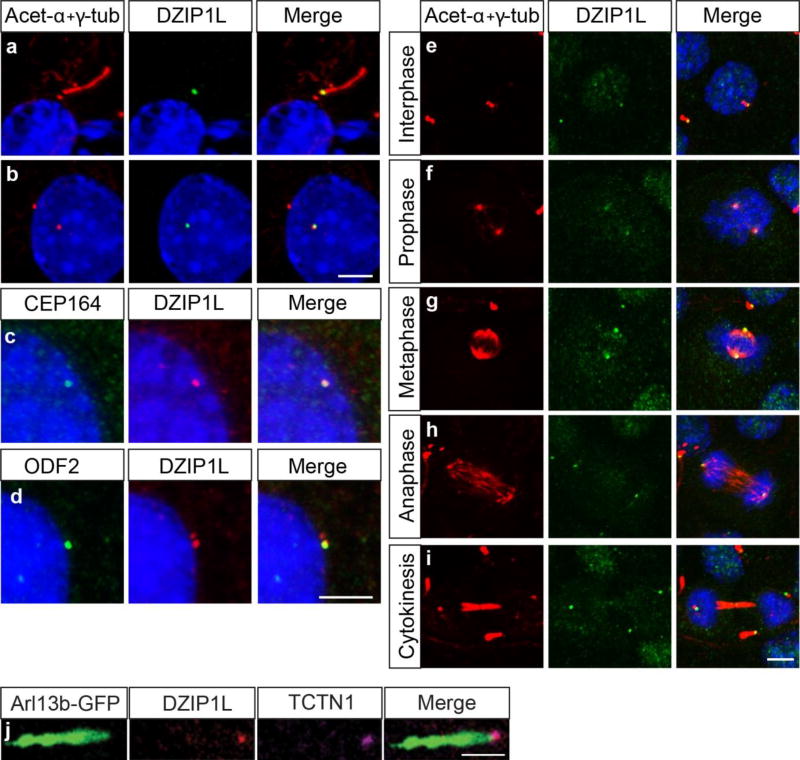
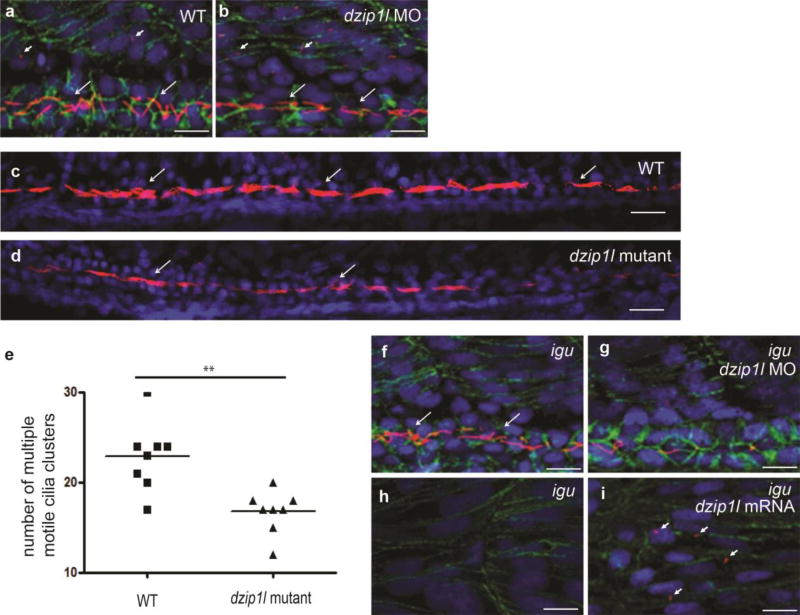
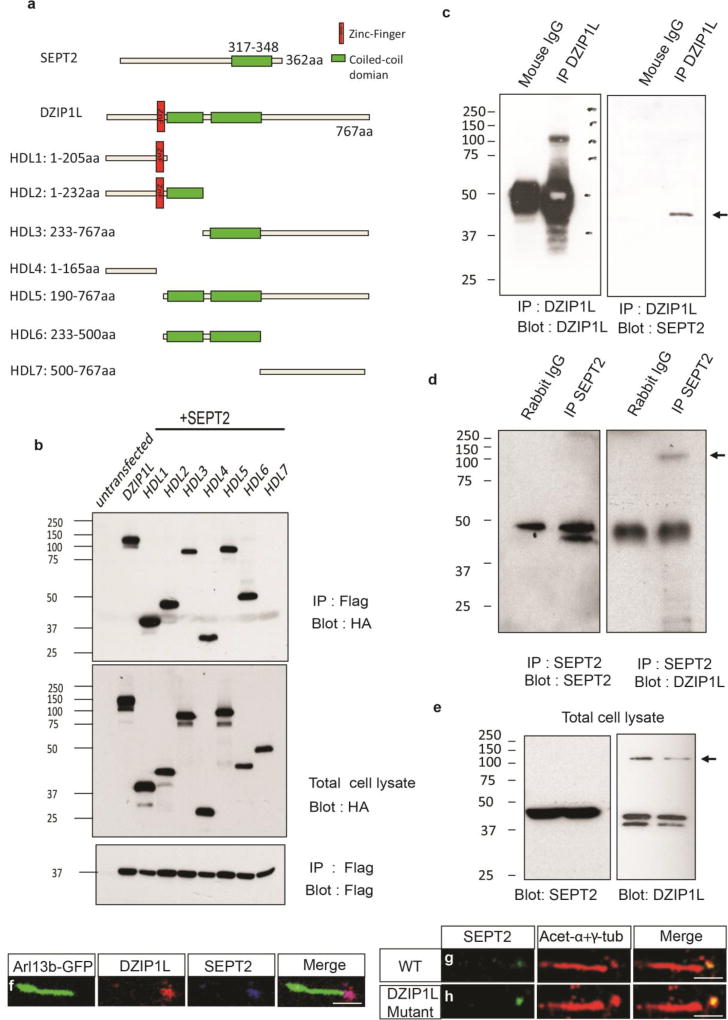
Autosomal recessive polycystic kidney disease (ARPKD), usually considered to be a genetically homogeneous disease caused by mutations in PKHD1, has been associated with ciliary dysfunction. Here, we describe mutations in DZIP1L, which encodes DAZ interacting protein 1-like, in patients with ARPKD. We further validated these findings through loss-of-function studies in mice and zebrafish. DZIP1L localizes to centrioles and to the distal ends of basal bodies, and interacts with septin2, a protein implicated in maintenance of the periciliary diffusion barrier at the ciliary transition zone. In agreement with a defect in the diffusion barrier, we found that the ciliary-membrane translocation of the PKD proteins polycystin-1 and polycystin-2 is compromised in DZIP1L-mutant cells. Together, these data provide what is, to our knowledge, the first conclusive evidence that ARPKD is not a homogeneous disorder and further establish DZIP1L as a second gene involved in ARPKD pathogenesis.
Figures
References
-
- Bergmann C, et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD) Kidney Int. 2005;67:829–48. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
