

A Role for the Host DNA Damage Response in Hepatitis B Virus cccDNA Formation-and Beyond?
- PMID: 28531167
- PMCID: PMC5454437
- DOI: 10.3390/v9050125
A Role for the Host DNA Damage Response in Hepatitis B Virus cccDNA Formation-and Beyond?
Abstract
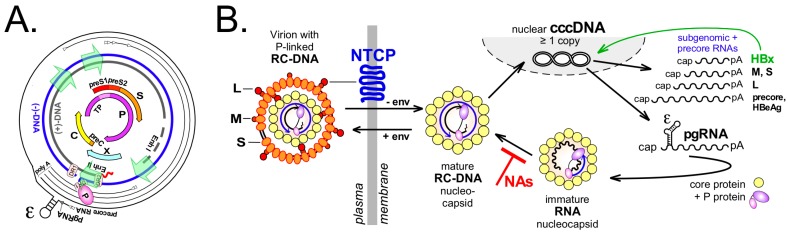
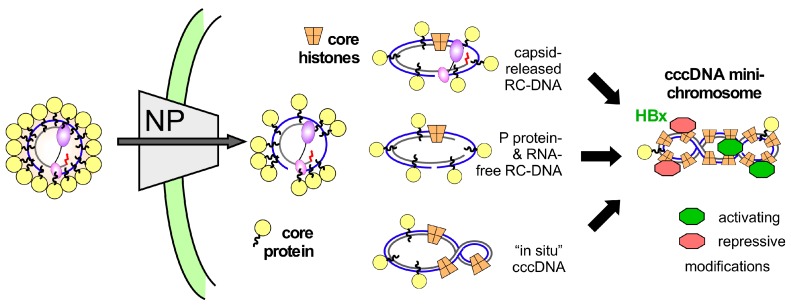
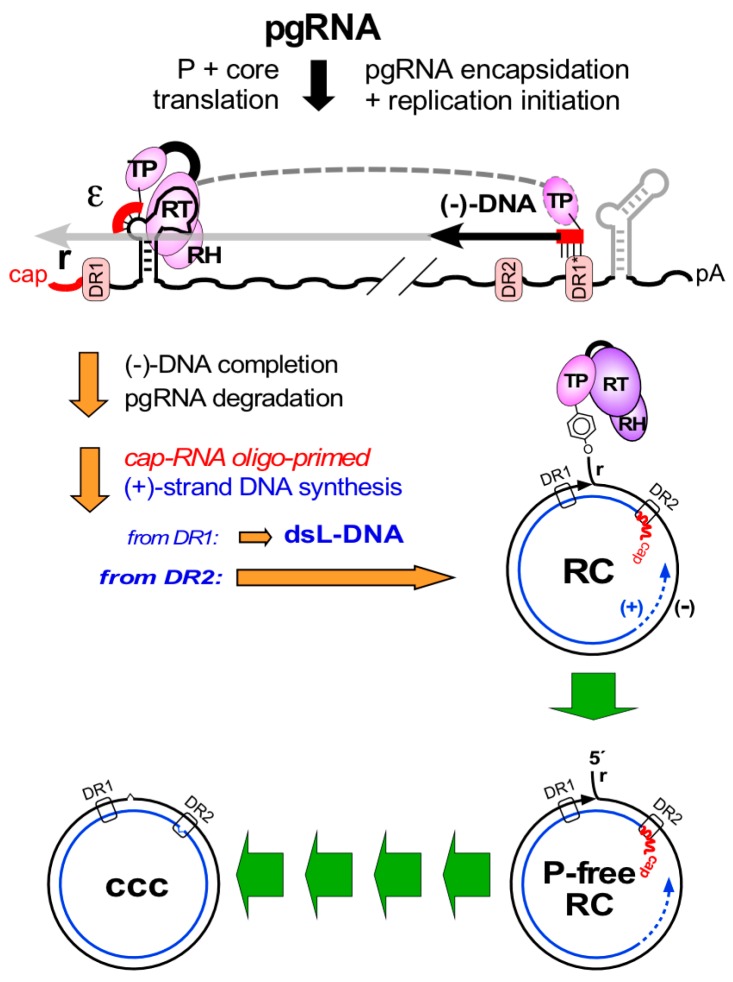
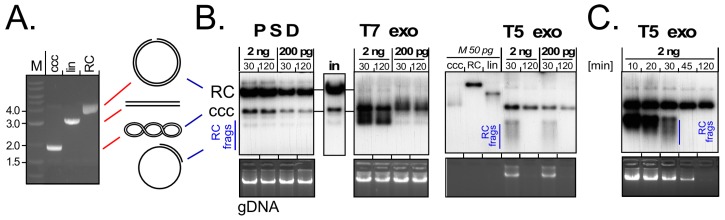
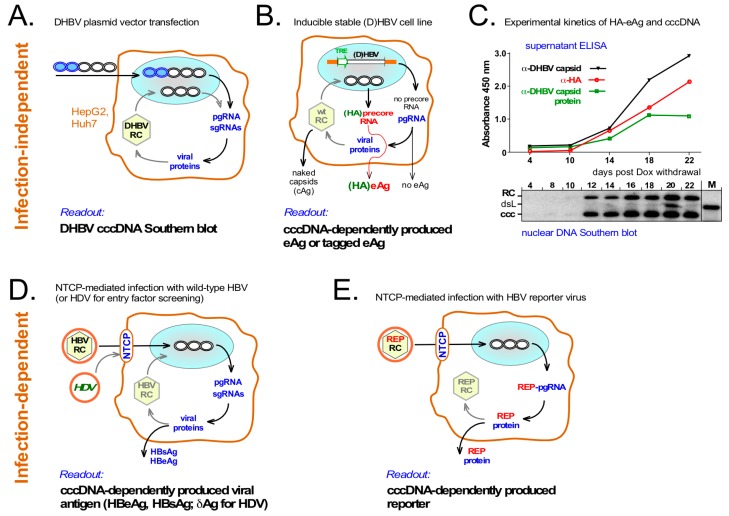
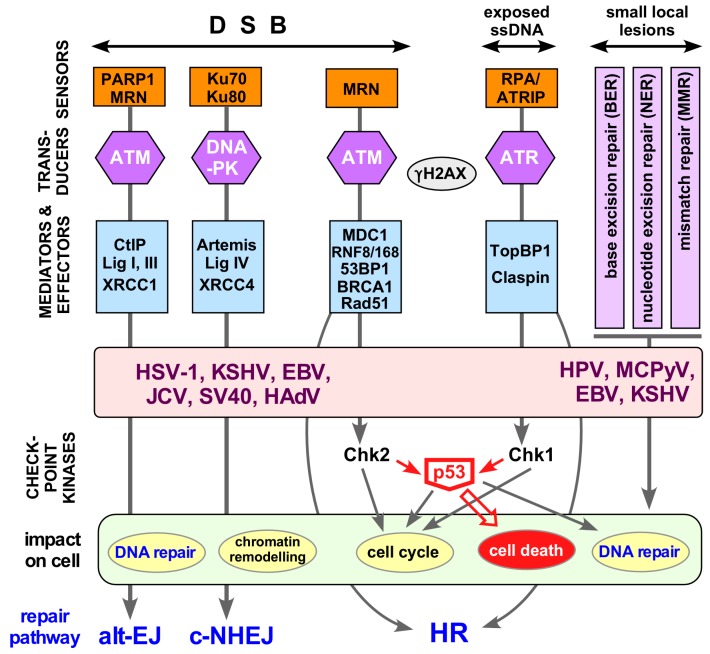
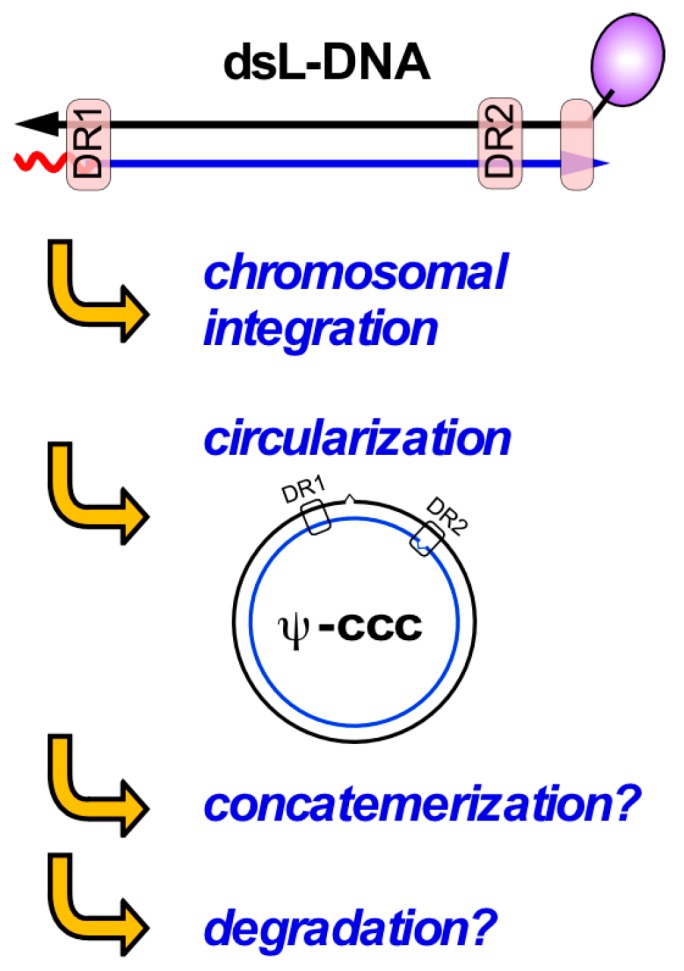
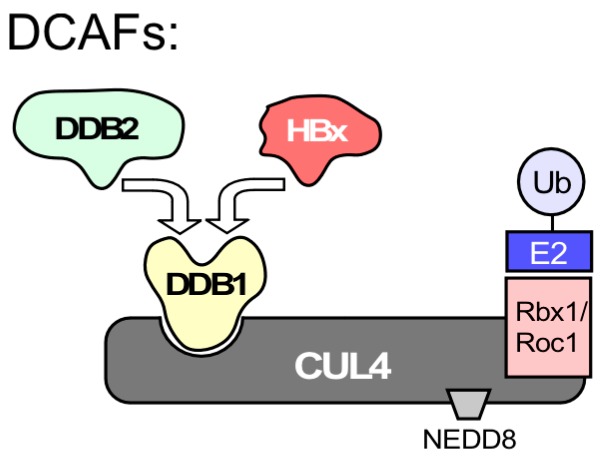
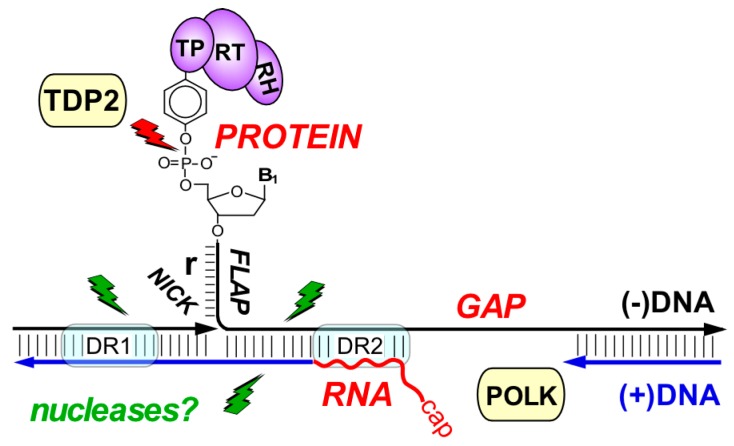
Chronic hepatitis B virus (HBV) infection puts more than 250 million people at a greatly increased risk to develop end-stage liver disease. Like all hepadnaviruses, HBV replicates via protein-primed reverse transcription of a pregenomic (pg) RNA, yielding an unusually structured, viral polymerase-linked relaxed-circular (RC) DNA as genome in infectious particles. Upon infection, RC-DNA is converted into nuclear covalently closed circular (ccc) DNA. Associating with cellular proteins into an episomal minichromosome, cccDNA acts as template for new viral RNAs, ensuring formation of progeny virions. Hence, cccDNA represents the viral persistence reservoir that is not directly targeted by current anti-HBV therapeutics. Eliminating cccDNA will thus be at the heart of a cure for chronic hepatitis B. The low production of HBV cccDNA in most experimental models and the associated problems in reliable cccDNA quantitation have long hampered a deeper understanding of cccDNA molecular biology. Recent advancements including cccDNA-dependent cell culture systems have begun to identify select host DNA repair enzymes that HBV usurps for RC-DNA to cccDNA conversion. While this list is bound to grow, it may represent just one facet of a broader interaction with the cellular DNA damage response (DDR), a network of pathways that sense and repair aberrant DNA structures and in the process profoundly affect the cell cycle, up to inducing cell death if repair fails. Given the divergent interactions between other viruses and the DDR it will be intriguing to see how HBV copes with this multipronged host system.
Keywords: DNA damage response; DNA repair; HBV cure; HBV minichromosome; cccDNA; hepatitis B virus.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Stanaway J.D., Flaxman A.D., Naghavi M., Fitzmaurice C., Vos T., Abubakar I., Abu-Raddad L.J., Assadi R., Bhala N., Cowie B., et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet. 2016;388:1081–1088. doi: 10.1016/S0140-6736(16)30579-7. - DOI - PMC - PubMed
-
- WHO Fact sheet 297 Cancer 2017. Feb, 2017.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
