

Changes in the dynamics of the cardiac troponin C molecule explain the effects of Ca2+-sensitizing mutations
- PMID: 28533433
- PMCID: PMC5512083
- DOI: 10.1074/jbc.M116.770776
Changes in the dynamics of the cardiac troponin C molecule explain the effects of Ca2+-sensitizing mutations
Abstract
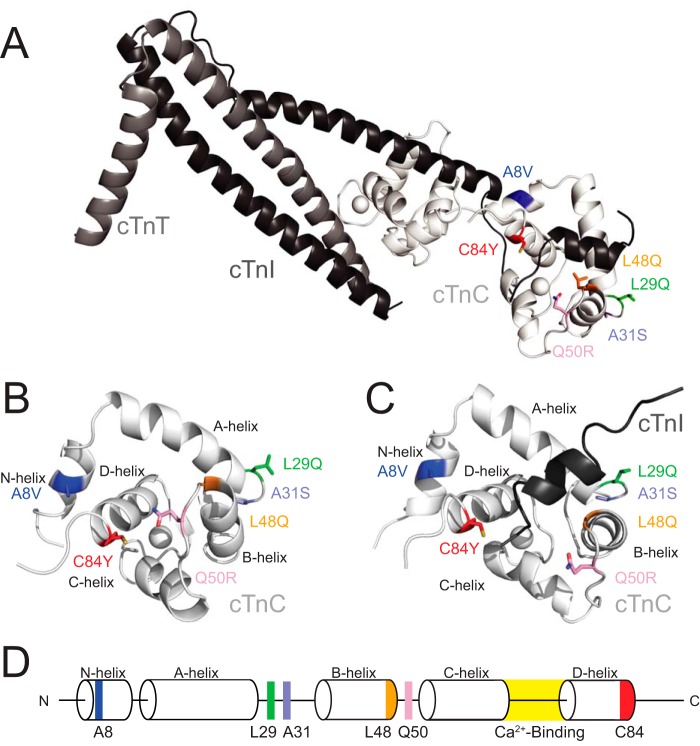
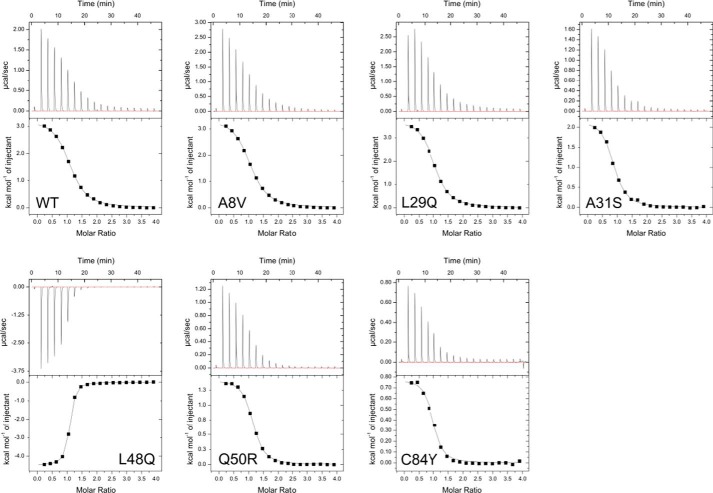
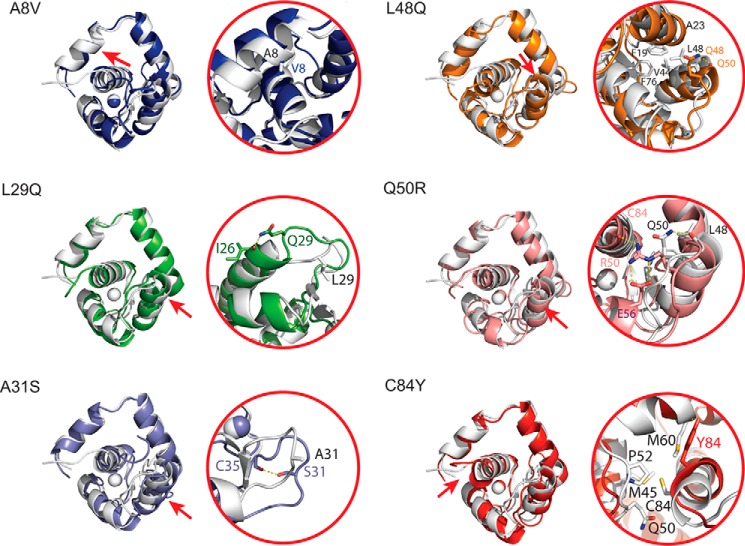
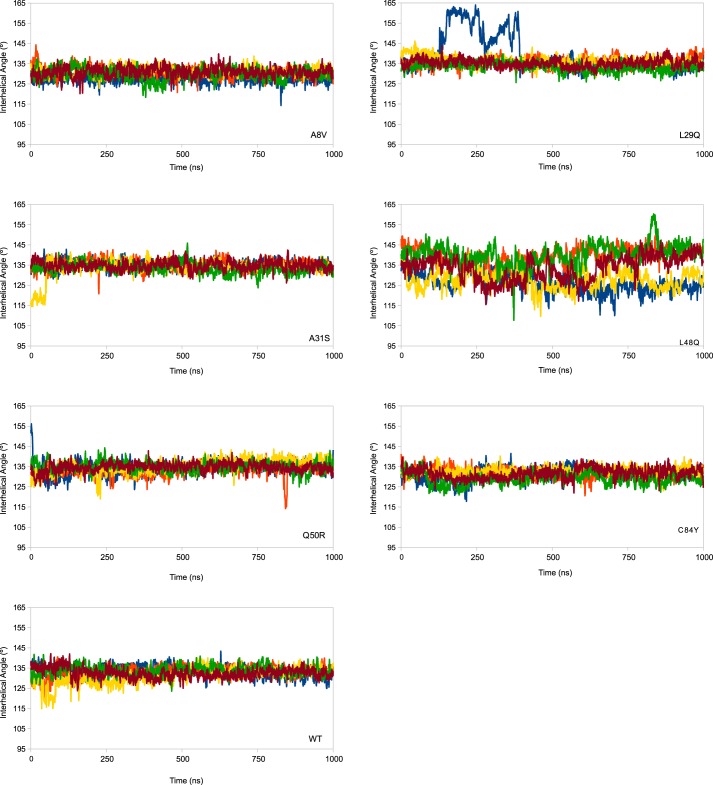
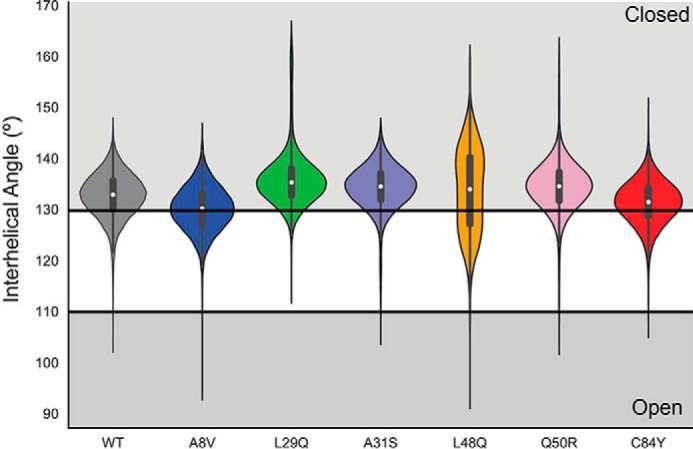
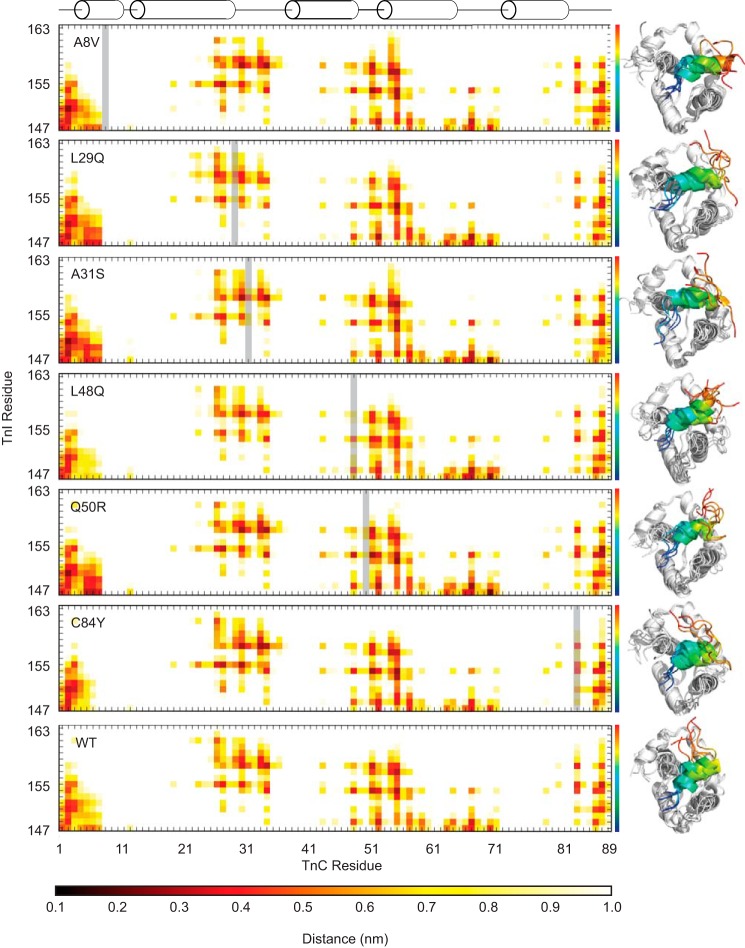
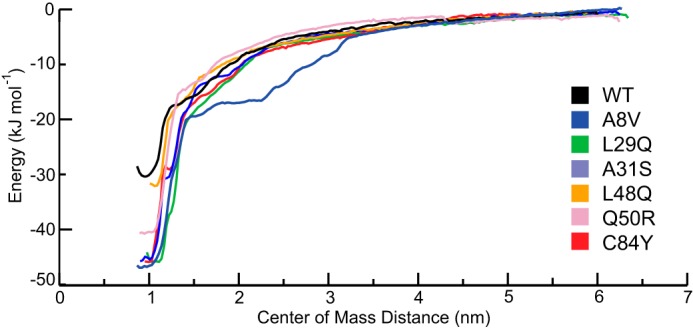
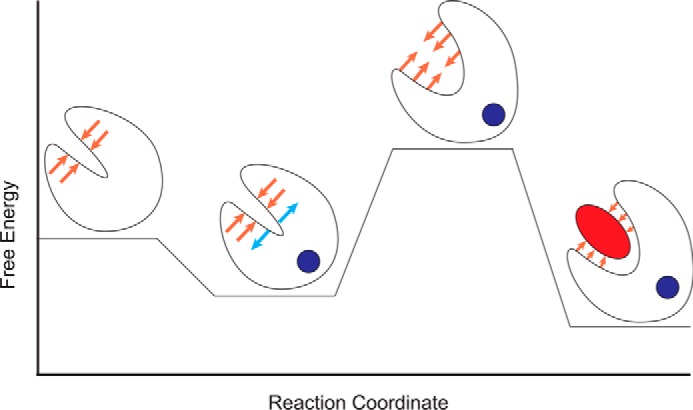
Cardiac troponin C (cTnC) is the regulatory protein that initiates cardiac contraction in response to Ca2+ TnC binding Ca2+ initiates a cascade of protein-protein interactions that begins with the opening of the N-terminal domain of cTnC, followed by cTnC binding the troponin I switch peptide (TnISW). We have evaluated, through isothermal titration calorimetry and molecular-dynamics simulation, the effect of several clinically relevant mutations (A8V, L29Q, A31S, L48Q, Q50R, and C84Y) on the Ca2+ affinity, structural dynamics, and calculated interaction strengths between cTnC and each of Ca2+ and TnISW Surprisingly the Ca2+ affinity measured by isothermal titration calorimetry was only significantly affected by half of these mutations including L48Q, which had a 10-fold higher affinity than WT, and the Q50R and C84Y mutants, each of which had affinities 3-fold higher than wild type. This suggests that Ca2+ affinity of the N-terminal domain of cTnC in isolation is insufficient to explain the pathogenicity of these mutations. Molecular-dynamics simulation was used to evaluate the effects of these mutations on Ca2+ binding, structural dynamics, and TnI interaction independently. Many of the mutations had a pronounced effect on the balance between the open and closed conformations of the TnC molecule, which provides an indirect mechanism for their pathogenic properties. Our data demonstrate that the structural dynamics of the cTnC molecule are key in determining myofilament Ca2+ sensitivity. Our data further suggest that modulation of the structural dynamics is the underlying molecular mechanism for many disease mutations that are far from the regulatory Ca2+-binding site of cTnC.
Keywords: calcium; cardiomyopathy; isothermal titration calorimetry (ITC); molecular dynamics; troponin.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Maron B. J., Shirani J., Poliac L. C., Mathenge R., Roberts W. C., and Mueller F. O. (1996) Sudden death in young competitive athletes: clinical, demographic, and pathological profiles. JAMA 276, 199–204 - PubMed
-
- Semsarian C., Ingles J., Maron M. S., and Maron B. J. (2015) New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 65, 1249–1254 - PubMed
-
- Harada K., and Morimoto S. (2004) Inherited cardiomyopathies as a troponin disease. Jpn. J. Physiol. 54, 307–318 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
