

Lid opening and conformational stability of T1 Lipase is mediated by increasing chain length polar solvents
- PMID: 28533982
- PMCID: PMC5438581
- DOI: 10.7717/peerj.3341
Lid opening and conformational stability of T1 Lipase is mediated by increasing chain length polar solvents
Abstract
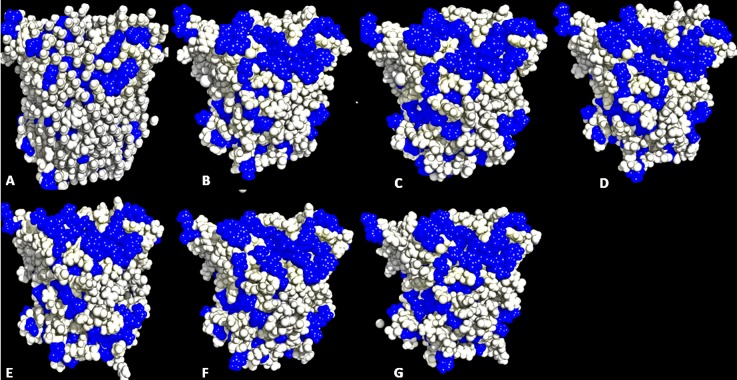
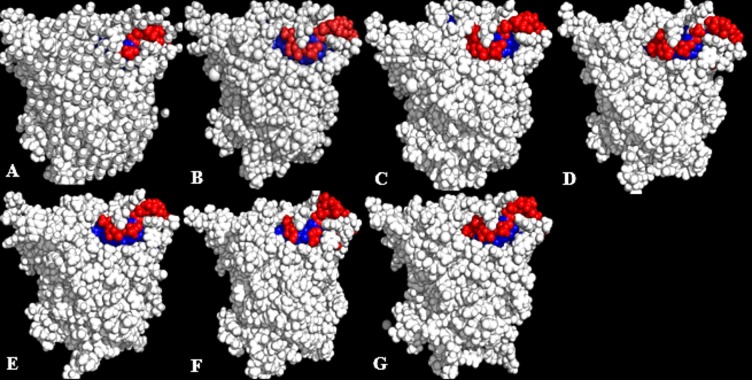
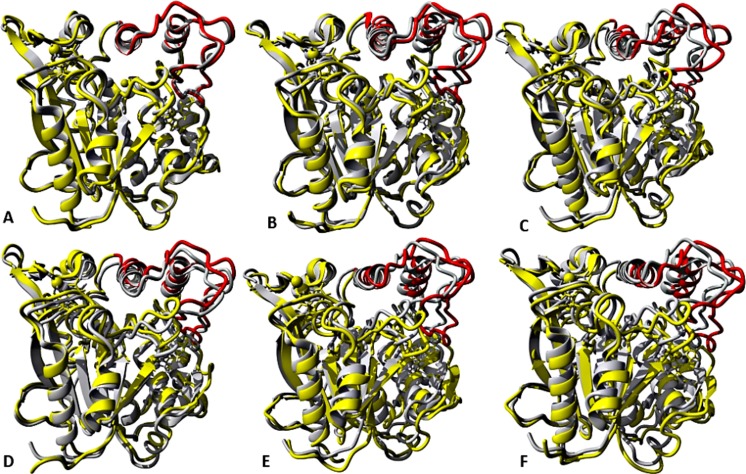
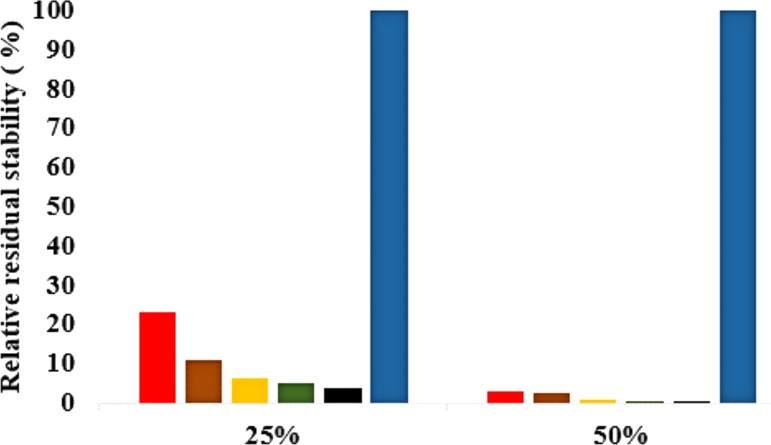
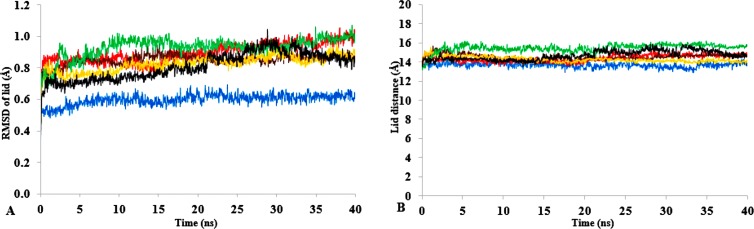
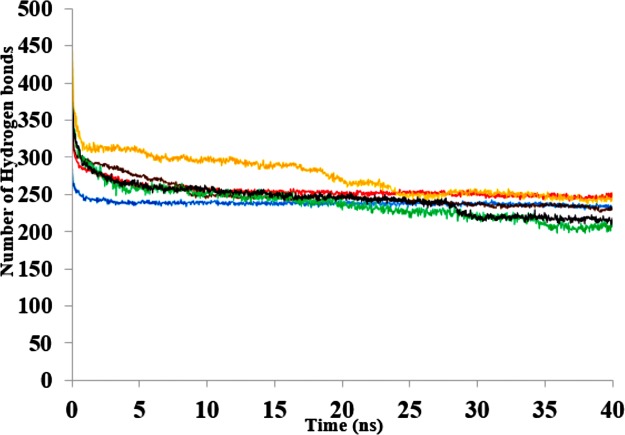
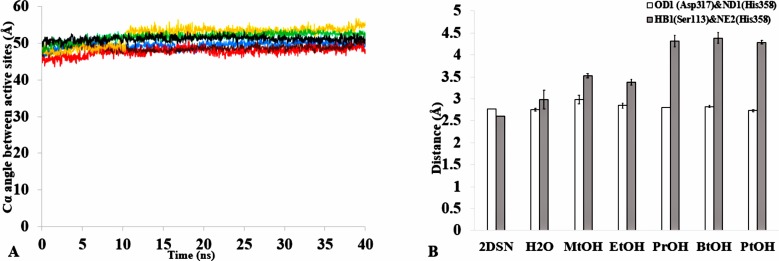
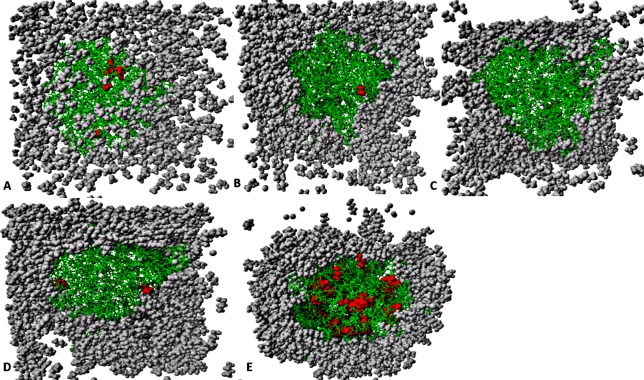
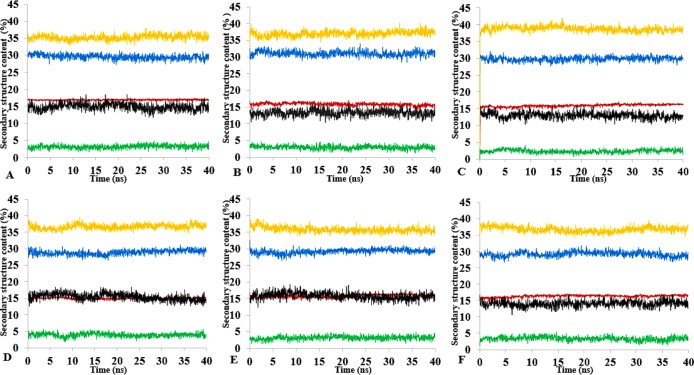
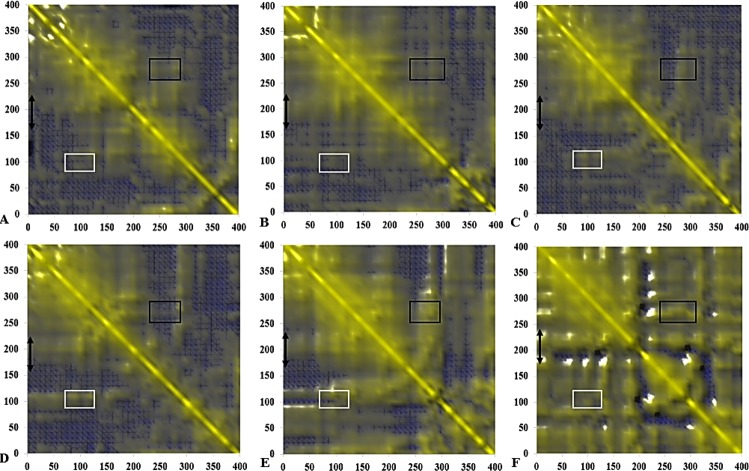
The dynamics and conformational landscape of proteins in organic solvents are events of potential interest in nonaqueous process catalysis. Conformational changes, folding transitions, and stability often correspond to structural rearrangements that alter contacts between solvent molecules and amino acid residues. However, in nonaqueous enzymology, organic solvents limit stability and further application of proteins. In the present study, molecular dynamics (MD) of a thermostable Geobacillus zalihae T1 lipase was performed in different chain length polar organic solvents (methanol, ethanol, propanol, butanol, and pentanol) and water mixture systems to a concentration of 50%. On the basis of the MD results, the structural deviations of the backbone atoms elucidated the dynamic effects of water/organic solvent mixtures on the equilibrium state of the protein simulations in decreasing solvent polarity. The results show that the solvent mixture gives rise to deviations in enzyme structure from the native one simulated in water. The drop in the flexibility in H2O, MtOH, EtOH and PrOH simulation mixtures shows that greater motions of residues were influenced in BtOH and PtOH simulation mixtures. Comparing the root mean square fluctuations value with the accessible solvent area (SASA) for every residue showed an almost correspondingly high SASA value of residues to high flexibility and low SASA value to low flexibility. The study further revealed that the organic solvents influenced the formation of more hydrogen bonds in MtOH, EtOH and PrOH and thus, it is assumed that increased intraprotein hydrogen bonding is ultimately correlated to the stability of the protein. However, the solvent accessibility analysis showed that in all solvent systems, hydrophobic residues were exposed and polar residues tended to be buried away from the solvent. Distance variation of the tetrahedral intermediate packing of the active pocket was not conserved in organic solvent systems, which could lead to weaknesses in the catalytic H-bond network and most likely a drop in catalytic activity. The conformational variation of the lid domain caused by the solvent molecules influenced its gradual opening. Formation of additional hydrogen bonds and hydrophobic interactions indicates that the contribution of the cooperative network of interactions could retain the stability of the protein in some solvent systems. Time-correlated atomic motions were used to characterize the correlations between the motions of the atoms from atomic coordinates. The resulting cross-correlation map revealed that the organic solvent mixtures performed functional, concerted, correlated motions in regions of residues of the lid domain to other residues. These observations suggest that varying lengths of polar organic solvents play a significant role in introducing dynamic conformational diversity in proteins in a decreasing order of polarity.
Keywords: Cross correlations; Interactions; Molecular dynamics; Organic solvent mixtures; Residue fluctuations; Structure conformation.
Conflict of interest statement
The authors declare there are no competing interests.
Figures
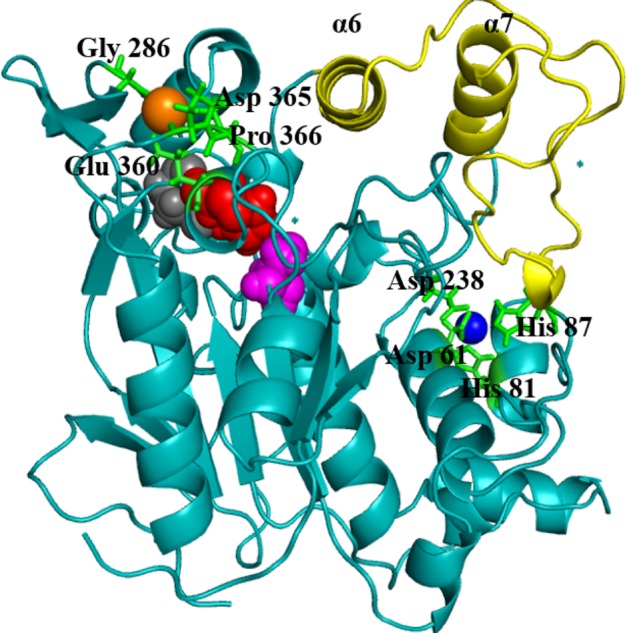
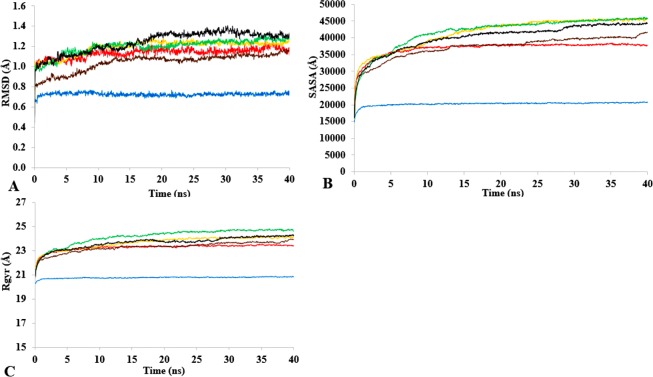
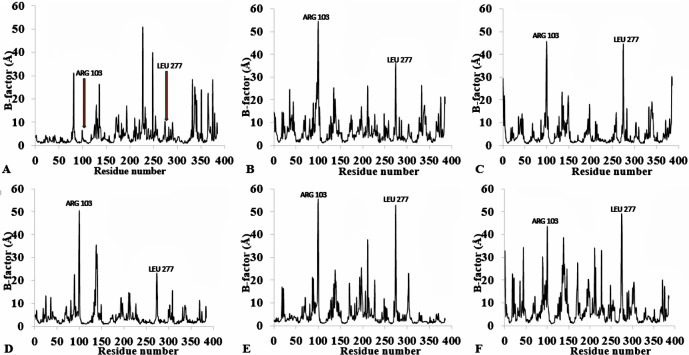
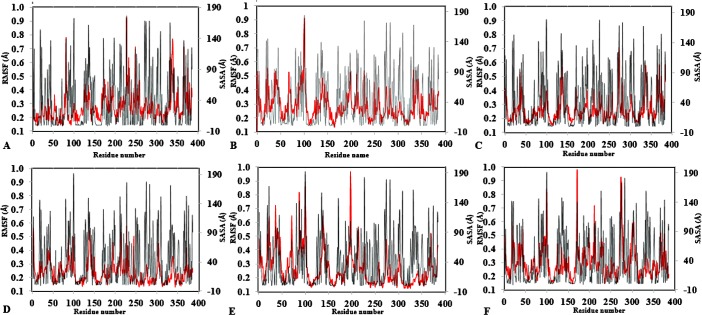
References
-
- Arcangeli C, Bizzarri AR, Cannistraro S. Molecular dynamics simulation and essential dynamics study of mutated plastocyanin: structural, dynamical and functional effects of a disulfide bridge insertion at the protein surface. Biophysical Chemistry. 2001;92:183–199. doi: 10.1016/S0301-4622(01)00199-5. - DOI - PubMed
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
